Resumos
Foram utilizados diferentes níveis de significância genômica na seleção assistida por marcadores para estimar a endogamia média e o limite de seleção, assim como os valores fenotípicos, em características quantitativas de baixa, média e alta herdabilidade. Uma comparação entre os níveis de 1%, 5%, 10% e 20% foi realizada por meio do sistema computacional de simulação genética (GENESYS), utilizado para a simulação de três genomas (cada qual constituído de um único caráter de baixa, média ou alta herdabilidade), e das populações base e inicial. Os resultados indicaram superioridade dos níveis de significância de maior magnitude (10% e 20%) com relação aos valores fenotípicos, resultante de menor média endogâmica, além de menor limite de seleção ao longo das gerações sob seleção para estes níveis. Estes resultados foram observados para todas as três características, embora de forma mais expressiva para o caráter de baixa herdabilidade. Assim, apesar de os níveis de 1% e 5% apresentarem maior precisão na detecção de marcadores ligados a quantitative trait loci (QTL), eles conduzem a maiores médias endogâmicas e limite de seleção, propiciando ganhos fenotípicos menores.
herdabilidade; nível de significância; simulação; QTL
Different levels of genomic significance were used in the selection assisted by molecular markers to estimate the medium endogamy and the selection limit, as well as the phenotypic value, for quantitative traits of low, medium, and high heritability. A comparison among the levels of genomic significance of 1%, 5%, 10%, and 20% was accomplished by a computer system of genetic simulation (GENESYS), used to simulate three genomes, each of them constituted by only one character of low, medium and high heritability, and to simulate the base and the initial populations. The results suggest superiority of higher significance levels (10% and 20%) for all phenotypic values, as a consequence of lower endogamy, and lower selection limit for low, medium, and high heritability traits, but in more expressive way for low heritability trait. Although the significant levels of 1% and 5% for molecular marker assisted selection showed a high precision in detecting markers related to a quantitative trait loci (QTLs), they lead to higher endogamy and selection limits, resulting in low phenotypic gains.
heritability; significance level; simulation; QTL
ZOOTECNIA E TECNOLOGIA E INSPEÇÃO DE PRODUTOS DE ORIGEM ANIMAL
Endogamia e limite de seleção em populações sob seleção assistida por marcadores moleculares
Endogamy and its selection limit in populations under assisted selection by molecular markers
M. JangarelliI, III; R.F. EuclydesII
IAluno de pós-graduação - UFV - Viçosa, MG
IIDepartamento de Zootecnia - UFV - Viçosa, MG
IIIBolsista do CNPq
RESUMO
Foram utilizados diferentes níveis de significância genômica na seleção assistida por marcadores para estimar a endogamia média e o limite de seleção, assim como os valores fenotípicos, em características quantitativas de baixa, média e alta herdabilidade. Uma comparação entre os níveis de 1%, 5%, 10% e 20% foi realizada por meio do sistema computacional de simulação genética (GENESYS), utilizado para a simulação de três genomas (cada qual constituído de um único caráter de baixa, média ou alta herdabilidade), e das populações base e inicial. Os resultados indicaram superioridade dos níveis de significância de maior magnitude (10% e 20%) com relação aos valores fenotípicos, resultante de menor média endogâmica, além de menor limite de seleção ao longo das gerações sob seleção para estes níveis. Estes resultados foram observados para todas as três características, embora de forma mais expressiva para o caráter de baixa herdabilidade. Assim, apesar de os níveis de 1% e 5% apresentarem maior precisão na detecção de marcadores ligados a quantitative trait loci (QTL), eles conduzem a maiores médias endogâmicas e limite de seleção, propiciando ganhos fenotípicos menores.
Palavras-chave: herdabilidade, nível de significância, simulação, QTL
ABSTRACT
Different levels of genomic significance were used in the selection assisted by molecular markers to estimate the medium endogamy and the selection limit, as well as the phenotypic value, for quantitative traits of low, medium, and high heritability. A comparison among the levels of genomic significance of 1%, 5%, 10%, and 20% was accomplished by a computer system of genetic simulation (GENESYS), used to simulate three genomes, each of them constituted by only one character of low, medium and high heritability, and to simulate the base and the initial populations. The results suggest superiority of higher significance levels (10% and 20%) for all phenotypic values, as a consequence of lower endogamy, and lower selection limit for low, medium, and high heritability traits, but in more expressive way for low heritability trait. Although the significant levels of 1% and 5% for molecular marker assisted selection showed a high precision in detecting markers related to a quantitative trait loci (QTLs), they lead to higher endogamy and selection limits, resulting in low phenotypic gains.
Keywords: heritability, significance level, simulation, QTL
INTRODUÇÃO
A disponibilidade de marcadores moleculares tem possibilitado diversos estudos na genética quantitativa. Para características quantitativas, vários marcadores são geralmente envolvidos (Saghai-Maroof et al., 1996; Dudley, 1997). QTLs têm o mesmo significado da herança quantitativa para o melhoramento clássico, ou seja, seu controle é condicionado por vários genes, e o fenótipo dos indivíduos é definido em termos de variâncias. O propósito do mapeamento de QTLs é inferir sobre os seus genótipos, objetivando estimar seus efeitos e sua localização, a partir de associações estatísticas com marcadores genéticos conhecidos (Liu, 1998).
Os marcadores moleculares são seqüências de DNA e podem ser identificados e mapeados, sendo utilizados para identificar e localizar genes específicos nos cromossomos (Anderson et al., 1994). A seleção assistida por marcadores moleculares (SAMM) é condicionada pela significância genômica exigida nas associações entre os genótipos dos marcadores e os QTLs, que são regiões cromossômicas relacionadas com a variação fenotípica das características quantitativas (Hayward et al., 1994). O uso dos marcadores em modernos programas de melhoramento baseia-se no princípio de que, se um gene ou conjunto de genes está associado a um marcador molecular de fácil identificação, então a seleção para este marcador será mais eficiente do que para a própria característica (Hayward et al., 1994). A SAMM consiste de dois passos principais: identificação de associações entre locos marcadores e QTLs, e o uso destas associações para o desenvolvimento de populações melhoradas (Bulfield, 1997). Entretanto, a SAMM deve ser encarada como uma ferramenta auxiliar e não substituta dos métodos tradicionais de melhoramento (Lande e Thompson, 1990).
A capacidade de detecção de associações entre marcadores - QTL é limitada pelo tamanho da população, pela exigência estatística para detecção dos locos (nível de significância genômica), pelo número de marcadores utilizados no mapeamento e pela herdabilidade da característica em estudo (Liu, 1998).
Como poucos locos economicamente importantes são conhecidos, e os QTLs não têm suas bases moleculares definidas, esses atualmente têm sido referidos mais como uma associação estatística do que como uma entidade biológica, sendo a herança acompanhada pelos marcadores (Guimarães, 2004). Portanto, os QTLs têm sido identificados como associações estatísticas entre dados relativos a uma região genômica e a variabilidade fenotípica existente entre populações segregantes (Li, 1998). Por serem assim caracterizados, os procedimentos para sua localização e estimação são tipicamente baseados em testes estatísticos (Burrow e Blake, 1998), identificando marcadores mais fortemente associados com o surgimento de um determinado fenótipo (William, 1998).
Este trabalho teve o objetivo de avaliar diferentes níveis de significância genômica, utilizados na SAMM, com relação aos parâmetros genéticos endogamia e limite de seleção, assim como seus efeitos sobre o valor fenotípico de características quantitativas de baixa, média e alta herdabilidade.
MATERIAL E MÉTODOS
Os dados utilizados neste trabalho foram simulados via sistema computacional de simulação genética - GENESYS (versão 2007), desenvolvido por Euclydes (1996). Este sistema é escrito na linguagem de programação FORTRAN, permitindo a criação de genomas de certa complexidade, que podem ser utilizados para a formação de populações de acordo com a estrutura desejada, sob influência de questionamentos propostos a serem analisados, seja por meio de métodos de seleção, pressuposições estatísticas, sistemas de acasalamentos, dentre outros fatores, dispensando, portanto, animais e laboratórios.
Para o presente estudo, foram simulados genomas semelhantes ao de frangos de corte Gallus gallus, possibilitando estudar os efeitos de diferentes níveis de significância, admitidos na SAMM, em características de baixa, média e alta herdabilidade, sobre os seguintes parâmetros: endogamia e limite de seleção, além do efeito desses parâmetros nos valores fenotípicos.
Utilizando o programa GENESYS, foram simulados três genomas, cada qual contendo 80 marcadores moleculares. Cada genoma era constituído de uma única característica quantitativa com herdabilidade de 0,10, 0,35 ou 0,60, respectivamente, designada simplesmente como peso (kg), governada por 200 locos quantitativos (QTLs).
Todos os genomas simulados estavam assim caracterizados geneticamente: genoma com 958 centimorgans de comprimento; 40 cromossomos de tamanho aleatório; os efeitos aditivos dos locos quantitativos foram simulados, segundo a distribuição normal; os locos quantitativos foram dialélicos e não possuíam desvios de dominância nem interações entre si; não possuíam cromossoma sexual e as freqüências gênicas iniciais eram iguais para ambos os sexos; as freqüências gênicas iniciais para os locos quantitativos seguiam distribuição normal, apresentando valores próximos a 0,5; as freqüências gênicas iniciais para os marcadores moleculares seguiam distribuição uniforme, apresentando valores próximos a 0,5; os efeitos de ambiente foram simulados, conforme a distribuição normal; apresentava uma média e um desvio-padrão fenotípico de 2,00kg e 0,20kg, respectivamente.
Para cada estrutura genômica simulada, foi construída uma população base, composta de 500 machos e 500 fêmeas, não aparentados. Com os 1000 descendentes escolhidos aleatoriamente em cada população base, obtidos do cruzamento de 10 machos e 10 fêmeas/macho (100 fêmeas), produzindo 10 filhos/fêmea/macho (1000 indivíduos), formaram-se as populações iniciais que, por sua vez, foram submetidas à seleção assistida por marcadores moleculares por vinte gerações consecutivas com 10 repetições, visando minimizar os efeitos da flutuação genética. A partir de cada uma dessas três populações iniciais, os reprodutores eram selecionados com base nos genótipos de um número de marcadores moleculares, que estariam estatisticamente associados a locos quantitativos. A cada geração, os 10 machos e as 100 fêmeas (10 fêmeas/macho) que obtiveram os melhores desempenhos foram acasalados ao acaso, produzindo 1000 descendentes (10 filhos por acasalamento) que, por sua vez, formavam a geração seguinte.
Dessa forma, obtida a população inicial para cada genoma, foram praticadas quatro seleções assistida por marcadores, com diferentes níveis de significância genômicas adotados para a identificação de marcadores associados aos QTLs de interesse, todas partindo do mesmo valor fenotípico, sendo que a identificação dava-se por meio de uma análise de regressão linear simples entre os genótipos dos marcadores com os valores fenotípicos dos descendentes dos acasalamentos.
Os níveis de significância que caracterizavam os quatro processos de SAMM, por genoma/ população inicial, foram: nível de significância de 1%, considerado de alta significância, que detecta somente os marcadores que estiverem mais fortemente relacionados ao genótipo dos QTLs e, conseqüentemente, ao fenótipo da característica de interesse; ele apresenta maior precisão, contudo, resulta em menor número de marcadores identificados na seleção. Nível de significância de 5%, significativo, que seleciona os marcadores - QTLs altamente significativos ao nível de 1%, assim como os significativos para maiores valores de significância genômica, ou seja, para 2%, 3%, 4% ou até 5%; dessa forma, um número igual ou maior de marcadores será selecionado, em comparação ao nível de 1%, em função da menor precisão exigida. Nível de significância de 10%, os marcadores - QTLs selecionados a este nível são referidos como sugestivos, tornam-se indicadores de possíveis associações entre seu genótipo e as expressões fenotípicas nos indivíduos; um maior número de marcadores será selecionado, compreendendo os altamente significativos (α = 1%), os significativos (α = 5%) e os sugestivos (α entre 5% e 10%). Nível de significância de 20%, os marcadores - QTLs selecionados também serão referidos como sugestivos, porém, são menos preferidos que os sugestivos no nível de 10% em razão do maior erro a ele associado (20%). Um número igual ou mais alto de marcadores será selecionado, em comparação com os níveis anteriores, pois representa a soma dos que foram significativos aos níveis de 1%, 5% e 10%, disponibilizando maior número de informações, contudo, com menor precisão.
RESULTADOS E DISCUSSÃO
Na Fig. 1, são apresentados os coeficientes médios de endogamia no decorrer das 20 gerações sob SAMM, para características de baixa herdabilidade, h2=0,10.
Em todos os quatro processos seletivos, observaram-se aumentos no coeficiente de endogamia, conduzindo a valores elevados nas últimas gerações, sendo que, ao praticar seleção contínua sobre uma população, os coeficientes endogâmicos tendem a convergir para valores bem próximos de 1,0 (Belonsky e Kennedy, 1988; Kuhlers e Kennedy, 1992; Quinton et al., 1992; Jeyruban et al., 1995; Quinton e Smith, 1995). É digno de nota lembrar que a endogamia tende a fixar alelos favoráveis, assim como desfavoráveis, em alguns locos (Breda et al., 2004). Dessa forma, os processos seletivos que conduzem a maiores médias endogâmicas favorecem a redução da variabilidade genética nas populações.
Ao trabalhar com níveis de significância de menor magnitude, o poder de detecção exigido nas associações entre o genótipo do marcador e a expressão fenotípica do caráter poderá afetar a variabilidade genética da população (Darvasi et al., 1993). Ao adotar alta significância na SAMM, favorecerá a uniformidade genética entre os animais, pois somente os que apresentam respectivos genes (QTLs), de maior efeito ou mais próximos dos marcadores utilizados, serão selecionados, propiciando a ocorrência de maiores coeficientes de endogamia (acasalamento entre indivíduos aparentados).
Os valores referentes ao limite de seleção são apresentados na Fig. 2. A redução de forma mais expressiva é prejudicial, resultando em menores ganhos fenotípicos a serem alcançados nas gerações seguintes sob seleção, conforme mostra a Fig 3, visto que um decréscimo mais acentuado nos processos seletivos, evidenciados na Fig. 2, representa, em média, maior limite de seleção (em kg) disponível à técnica seletiva.
De maneira geral, em todas as quatro seleções, foram observados decréscimos quanto aos valores fenotípicos limites a serem selecionados, ou seja, aumento no parâmetro limite de seleção. De acordo com Falconer (1987), não se pode esperar respostas à seleção de forma contínua, sendo que, após um determinado número de gerações sob seleção, todos os locos favoráveis tendem a ser fixados. À medida que os locos se aproximam da fixação, há redução na resposta à seleção, de maneira que, ao cessar a resposta, a população é dita estar no limite de seleção (Breda et al., 2004).
A adoção de níveis de alta significância favorecerá o aumento da endogamia (Fig. 1), resultando em maiores perdas na variância genética, o que torna, conseqüentemente, mais lenta a resposta à seleção ao longo das gerações, minimizando os ganhos fenotípicos a serem alcançados devido à maximização do limite de seleção, em comparação aos demais níveis (Fig. 2 e 3).
Em contrapartida, níveis sugestivos de significâncias aumentam o limite da seleção (redução em kg) de forma mais gradativa, proporcionando maiores ganhos fenotípicos com a seleção, devido ao menor aumento médio na endogamia.
Nas Fig. 4, 5 e 6 são apresentados os coeficientes médios de endogamia limite de seleção e valores fenotípicos, respectivamente de média herdabilidade, h2 = 0,35. Novamente, aumentos foram observados, porém com menor diferenciação entre os níveis, pois, à medida que o caráter torna-se mais herdável, a eficiência da SAMM se reduz e, conseqüentemente, menor diferenciação entre os níveis de significância utilizados na seleção será obtida (Lande e Thompson, 1990).
Os níveis de alta significância prevaleceram, conforme obtido para o caráter de baixa herdabilidade, no aumento da endogamia e do limite de seleção, assim como no menor valor fenotípico obtido ao longo das gerações, contudo, as diferenças entre os níveis significativos e os sugestivos adotados tornaram-se menores (Fig. 4, 5 e 6).
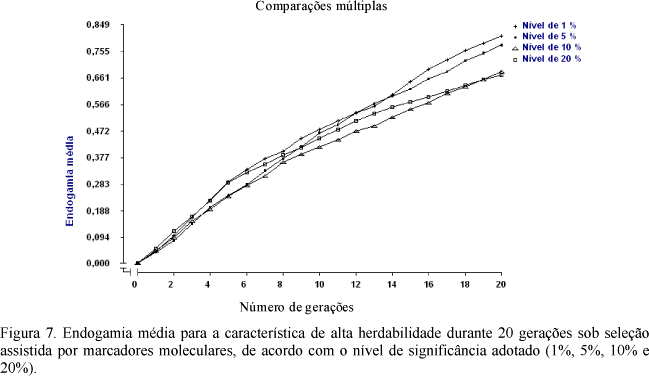
Os coeficientes médios de endogamia, limite de seleção e valores fenotípicos para características de alta herdabilidade, h2 = 0,60, são apresentados nas Fig. 7, 8 e 9, respectivamente. Mais uma vez, observou-se superioridade dos níveis de alta significância, no limite de seleção e no coeficiente endogâmico, assim como menores valores fenotípicos. Tal superioridade apresenta menor amplitude, em comparação às características anteriores, confirmando a menor eficiência desse processo seletivo (SAMM) para características de alta herdabilidade, de acordo com o comentário feito por Lande e Thompson (1990).
CONCLUSÕES
Adotando diferentes níveis de significância genômica na SAMM, obtiveram-se coeficientes endogâmicos e limites seletivos distintos como resposta à seleção. Para os níveis de 1% e 5%, observaram-se maiores coeficientes endogâmicos e limites de seleção, minimizando os ganhos fenotípicos a serem obtidos. Para os níveis de 10% e 20%, menores médias endogâmicas e limites de seleção foram observadas, resultando, conseqüentemente, em melhores respostas fenotípicas. Ao se relacionar resposta fenotípica com herdabilidade da característica, observa-se maior distinção entre os níveis adotados nas características cuja SAMM apresenta maior eficiência, ou seja, características de baixa herdabilidade. Para características de média e alta herdabilidade, a relevância de estabelecer os níveis adequados para detectar associações entre marcadores e QTLs se reduz, em função da menor diferenciação nos resultados, seja admitindo níveis significativos ou sugestivos.
Recebido em 1 de fevereiro de 2008
Aceito em 12 de novembro de 2008
E-mail: gmejanga@hotmail.com
- ANDERSON, L.; HALEY, C.S.; ELLEGREN, H. et al. Genetic mapping of quantitative trait loci for growth and fatness in pigs. Science, v.263, p.1771-1774, 1994.
- BELONSKY, G.M.; KENNEDY, B.W. Selection on individual phenotype and best linear unbiased predictor of breeding value in a closed swine herd. J. Anim. Sci., v.66, p.1124-1131, 1988.
- BREDA, F.C.; EUCLYDES, R.F.; PEREIRA, C.S. et al. Endogamia e limite de seleção em populações selecionadas obtidas por simulação. Rev. Bras. Zootec, v.33, p.2017-2025, 2004.
- BULFIELD, G. Strategies for the future. Poult. Sci., v.76, p.1071-1074, 1997.
- BURROW, M.D.; BLAKE T.K. Molecular tools for the study of complex traits. In: ___________Molecular dissection of complex traits New York: CRC, 1998. p.13-30.
- DARVASI, A.; WEINREB, V.; MINKE, V. et al. Detecting Marker-QTL Linkage and Estimating QTL Gene Effect and Map Location Using a Satured Genetic Map. Genetics, v.134, p.943-951, 1993.
- DUDLEY, J.W. Quantitative genetics and plant breeding. Adv. Agron., v.59, p.1-23, 1997.
- EUCLYDES, R.F. Uso do sistema para simulação Genesys na avaliação de métodos de seleção clássicos e associados a marcadores moleculares 1996. 149f. Tese (Doutorado) - Universidade Federal de Viçosa, Viçosa.
- FALCONER, D. S. Introdução à genética quantitativa Viçosa: UFV, 1987. 279p.
- GUIMARAES, S.E.F. Análise de marcadores genômicos e detecção de QTLs e genes candidatos em melhoramento animal. In: PEREIRA, J.C.C. Melhoramento genético aplicado à produção animal 4.ed. Belo Horizonte: FEPMVZ, 2004. p.491-524.
- HAYWARD, M.D.; McADAM, N.J.; JONES, J.G. et al. Genetic markers and the selection of quantitative traits in forage grasses. Euphytica, v.77, p.269-75, 1994.
- JEYARUBAN, M.G.; GIBSON, J.P.; GOWE, R.S. Comparison of index selection and best linear unbiased prediction for simulated layer poultry data. Poult. Sci., v.74, p.1566-1576, 1995.
- KUHLERS, D.L.; KENNEDY, B.W. Effect of culling on selection response using phenotypic selection or best linear unbiased prediction of breeding values in small, closed herds of swine. J. Anim. Sci., v.70, p.2338-2348, 1992.
- LANDE, R.; THOMPSON, R. Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics, v.124, p.743-756, 1990.
- LI, Z. Molecular analysis of epistasis. In: Molecular dissection of complex traits New York: CRC, 1998. p.119-130.
- LIU, B.H. Statistical genomics: linkage, mapping and QTL analysis Boca Raton: CRC, 1998. 611p.
- QUINTON, M.; SMITH, C.; GODDARD, M.E. Comparison of selection methods at the same level of inbreeding. J. Anim. Sci., v.70, p.1060-1067, 1992.
- QUINTON, M.; SMITH, C. Comparison of evaluation-selection system for maximizing genetic response at same level of inbreeding. J. Anim. Sci., v.73, p.2208-2212, 1995.
- SAGHAI-MAROOF, M.A.; YUE, Y.G.; XIANG, Z.X. et al. Identification of quantitative trait loci controlling resistance to gray leaf spot disease in maize. Theor. Appl. Gen., v.93, p.539-546, 1996.
- WILLIAM, D.B. QTL analysis: power, precision and accuracy. In: PATERSON, A.H. Molecular dissection of complex traits New York: CRC Press, 1998. p.145-162.
Datas de Publicação
-
Publicação nesta coleção
09 Fev 2009 -
Data do Fascículo
Dez 2008
Histórico
-
Aceito
12 Nov 2008 -
Recebido
01 Fev 2008