Resumos
O objetivo deste trabalho foi avaliar a eficiência do método de regressão em detectar QTL com base na utilização de dados da estrutura de família (irmãos completos e meios-irmãos), como aqueles gerados em um núcleo MOET. Foram simulados dados fenotípicos e genotípicos em uma estrutura de núcleo MOET fechado de seleção. Três arquivos foram analisados, contendo: a) informações conjuntas de irmãos completos e meios-irmãos; b) apenas informações de irmãos completos e c) apenas informações de meios-irmãos. Verificou-se que o método da regressão, para dados discretos ou contínuos, foi capaz de detectar associações entre marcador e QTL em níveis bastante expressivos de significância (P<0.001 e P<0,0001), quando se utilizou o arquivo que continha informações conjuntas de irmãos completos e meios-irmãos. Os resultados indicaram a possibilidade de utilização dessa metodologia para estudos de detecção/validação de QTL em rebanhos ou núcleos de seleção que utilizam MOET.
QTL; poder de detecção; marcador molecular; estrutura de família
The objective of this study was to evaluate the efficiency of the regression method to detect QTL using data from full and half-sib families, like those generated in a MOET nucleus. For this study, genotypic and phenotypic data were simulated in a structure of a closed selection MOET nucleus. Three files were analyzed containing: a) the joint information of full and half sibs; b) only full sibs data; and c) only half sibs data. The method of regression, for continuous or discrete data, was able to detect associations of marker and QTL in very expressive levels of significance (P<0.001 P<0.0001), when the file containing the joint information of full and half sisters was used. The results indicated the possibility of using this methodology for studies of QTL detection / validation in MOET nucleus or herds under selection.
QTL; power of detection; molecular marker; family structure
ZOOTECNIA E TECNOLOGIA E INSPEÇÃO DE PRODUTOS DE ORIGEM ANIMAL
Detecção de QTL em dados de famílias estruturadas como as de um núcleo MOET por meio do método da regressão
QTL detection in MOET nucleus family structure-like data using the regression method
M.G.C.D. PeixotoI; M.L. MartinezI; R.L. TeodoroI; M.A. MachadoI; M.R.S. CarvalhoII; F.C. GomesI; R.S. VernequeI
IEmbrapa Gado de Leite Rua Eugênio do Nascimento, 610 36038-330 - Juiz de Fora, MG
IIInstituto de Ciências Biológicas -UFMG - Belo Horizonte, MG
RESUMO
O objetivo deste trabalho foi avaliar a eficiência do método de regressão em detectar QTL com base na utilização de dados da estrutura de família (irmãos completos e meios-irmãos), como aqueles gerados em um núcleo MOET. Foram simulados dados fenotípicos e genotípicos em uma estrutura de núcleo MOET fechado de seleção. Três arquivos foram analisados, contendo: a) informações conjuntas de irmãos completos e meios-irmãos; b) apenas informações de irmãos completos e c) apenas informações de meios-irmãos. Verificou-se que o método da regressão, para dados discretos ou contínuos, foi capaz de detectar associações entre marcador e QTL em níveis bastante expressivos de significância (P<0.001 e P<0,0001), quando se utilizou o arquivo que continha informações conjuntas de irmãos completos e meios-irmãos. Os resultados indicaram a possibilidade de utilização dessa metodologia para estudos de detecção/validação de QTL em rebanhos ou núcleos de seleção que utilizam MOET.
Palavras-chave: QTL, poder de detecção, marcador molecular, estrutura de família
ABSTRACT
The objective of this study was to evaluate the efficiency of the regression method to detect QTL using data from full and half-sib families, like those generated in a MOET nucleus. For this study, genotypic and phenotypic data were simulated in a structure of a closed selection MOET nucleus. Three files were analyzed containing: a) the joint information of full and half sibs; b) only full sibs data; and c) only half sibs data. The method of regression, for continuous or discrete data, was able to detect associations of marker and QTL in very expressive levels of significance (P<0.001 P<0.0001), when the file containing the joint information of full and half sisters was used. The results indicated the possibility of using this methodology for studies of QTL detection / validation in MOET nucleus or herds under selection.
Keywords: QTL, power of detection, molecular marker, family structure
INTRODUÇÃO
A utilização de marcadores tem sido indicada como importante ferramenta para o monitoramento da diversidade genética, o planejamento de acasalamentos e a seleção assistida (Brascamp et al., 1993). Apesar de a eficiência da seleção assistida por marcadores não estar bem clara e documentada (Dekkers, 2004), tal seleção constitui uma das muitas estratégias propostas para aumentar o progresso genético em núcleos MOET de seleção, minimizando a endogamia.
A detecção de marcadores associados a QTL depende da estrutura de pedigree, da aferição sistemática das características, do número de indivíduos medidos, do grau de desequilíbrio de ligação marcador-QTL, do número de marcadores genotipados e do efeito do QTL sobre a característica (Martinez e Vukasinovic, 2000; Israel e Weller, 2002). Em animais de produção, o mapeamento de QTL por meio de marcador depende também da fonte de desequilíbrio e da história prévia da população.
Os primeiros métodos de detecção basearam-se na utilização de marcadores únicos, considerados efeitos fixos na análise de variância. Existem, entretanto, questionamentos sobre o poder de detecção e resolução de mapeamento por meio desses métodos. Outros delineamentos baseados no cruzamento entre linhagens divergentes (Simpson, 1989), cruzamentos entre populações segregantes (Beckmann e Soller, 1989) e estudos de família (Kashi et al., 1990) foram propostos para investigar a ligação marcador-QTL. O delineamento mais eficiente baseia-se na obtenção de linhagens endogâmicas divergentes, pois o desequilíbrio máximo de ligação é produzido na geração F1 do cruzamento entre as linhagens.
Uma série de metodologias foi desenvolvida, permitindo utilizar toda a informação disponível e maximizar o poder de detecção de QTL (Nicholas, 2006). Dentre estas: análise de informações de segregação incerta por meio do método da máxima verossimilhança (Fernando e Grossman, 1989), mapeamento de grupo de ligação em populações exogâmicas (Haley e Knott, 1992), mapeamento por intervalo para cruzamentos F2 e retrocruzamentos (Darvasi et al., 1993), delineamento de filhas, netas e bisnetas (Coppieters et al., 1999) e análise de informações de populações exogâmicas, por meio da amostragem de Gibbs e do método da Cadeia de Markov/Monte Carlo (Bink e van Arendonk, 1999; Bink et al., 2000).
Apesar da perda de poder de detecção, considerando que nem todos os indivíduos são informativos e a existência de diferenças individuais quanto à fase de ligação, a análise dentro de uma população exogâmica pode tornar evidentes as diferenças na segregação do QTL (Lynch e Walsh, 1998). Em gado de leite, usualmente, o desequilíbrio de ligação entre marcador e QTL pode ser encontrado dentro de famílias de meias-irmãs paternas, sendo que, quanto maior o número de filhas por reprodutor, maior o poder de detecção do QTL. Com vistas a aumentar ainda mais o poder de detecção, um algoritmo utilizando as informações de famílias de irmãs-completas e meias-irmãs paternas foi proposto (Le Roy et al., 1998).
No mapeamento de QTL por meio de marcadores, com quaisquer dos métodos ou delineamentos empregados, as informações fenotípicas sobre a característica quantitativa de interesse referem-se a irmãs completas ou a meiasirmãs (van der Beek et al., 1995). Esquemas de seleção em núcleos MOET têm sido implantados em alguns países (Strathie e McGuirk, 1995; Penna et al., 1998; Jokinen et al., 2003). Nesses esquemas, são produzidas famílias de irmãos completos e meios-irmãos, cujos dados produtivos são utilizados na avaliação genética dos animais. O objetivo deste estudo de simulação foi avaliar a eficiência de metodologias baseadas no método de regressão em detectar QTL com base na utilização de dados da estrutura de família (irmãos completos e meios-irmãos), como aqueles gerados em um núcleo MOET.
MATERIAL E MÉTODOS
O programa GENESYS foi utilizado para simulação dos dados genealógicos, fenotípicos e genotípicos, em uma estrutura populacional de um núcleo MOET (famílias de irmãos completos - IC/ meios-irmãos - MC), usando-se um modelo gênico (Euclydes, 1996). Os métodos numéricos desenvolvidos incluem procedimentos para mapear QTL em populações exogâmicas, com meios-irmãos e irmãos completos, usando-se múltiplos marcadores, para característica única.
Foram gerados 50 marcadores e 100 QTL, ambos dialélicos, distribuídos em cromossomos autossômicos, segundo regiões de consenso detectadas em estudos de mapeamento de QTL para características de produção de leite. Os efeitos genéticos aditivos e de meio foram normalmente distribuídos, de modo que a variância resultasse na herdabilidade de 0,29. Interações dos efeitos aditivo e não-aditivo foram ignoradas. Assumiram-se, inicialmente, frequências alélicas intermediárias (0,5) e uniformes em ambos os sexos.
A população-base do núcleo (cinco touros e 25 vacas) foi selecionada de 25 touros e 500 vacas. A cada geração, foram obtidos cinco irmãos completos por família e 125 filhos (irmãos completos e meios-irmãos) por touro, dos quais se selecionaram os pais da geração seguinte, evitando-se acasalamento entre indivíduos aparentados. Após 20 gerações, acumularam-se 2500 registros dos indivíduos, de seus respectivos pai e mãe, sexo, genótipos para 50 marcadores e de seu valor genético para a característica. Baseando-se nesse arquivo, dois outros foram aleatoriamente constituídos: um com dados de uma família por touro e apenas irmãos completos (IC), em um total de 100 indivíduos, e outro com apenas um filho por família e apenas meios-irmãos (MI) paternos, em um total de 500 indivíduos.
Para qualquer estrutura de família, a covariância entre os irmãos j e j' dentro da família i, assumindo-se endogamia nula, é igual a:
 em que:
em que:

 (irmãos completos)
(irmãos completos)
 (meios-irmãos),
(meios-irmãos),
sendo πq a proporção de alelos idênticos por descendência no loco candidato compartilhado pelos irmãos.
Para o estudo de associação, optou-se pelo método da regressão, para dados discretos (R) ou contínuos (AV), utilizando-se os seguintes arquivos, contendo diferentes fontes de informação: irmãos completos + meios-irmãos (IC+MI), apenas irmãos completos (IC) ou apenas meios-irmãos (MI). Foram realizadas análises de marcadores únicos, usando-se os procedimentos PROC REG, PROC GLM e PROC CORR do pacote computacional SAS/2003. O modelo fixo utilizado foi:
y= Xb + e, em que:
y= vetor de valores genéticos para a característica;
X= matriz de incidência relacionando os efeitos fixos dos marcadores aos valores genéticos;
b= vetor de efeitos fixos dos marcadores;
e= resíduos.
A significância da variação entre as classes genotípicas foi acessada pelo teste de F, e a dos efeitos aditivos associados a cada marcador, por meio de contrastes que compararam as médias das duas classes de homozigotos. Para se verificar o desvio de dominância, os contrastes compararam a média dos heterozigotos à média das classes de homozigotos. Para ambos, utilizou-se o teste t. Os efeitos de substituição gênica foram estimados por meio de contrastes específicos.
RESULTADOS E DISCUSSÃO
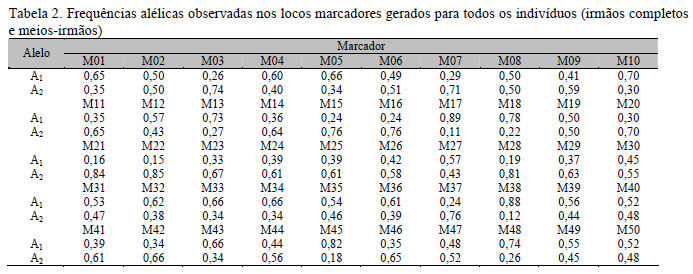
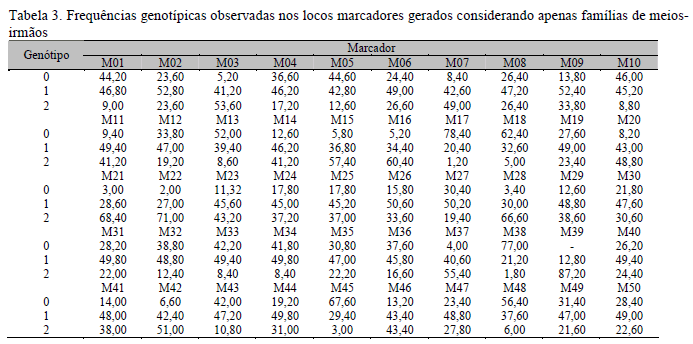
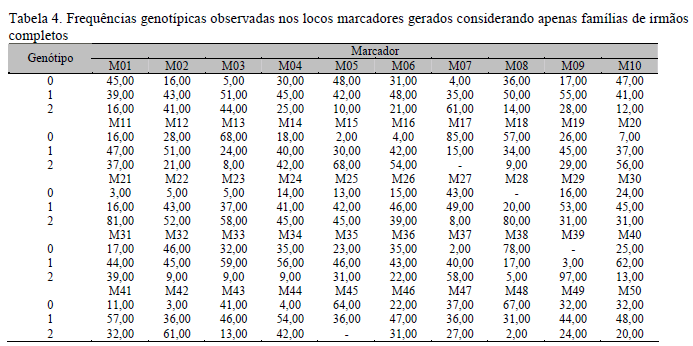
Nas Tab. 1, 2, 3 e 4 são apresentadas as frequências genotípicas e alélicas dos marcadores nos diferentes conjuntos de dados utilizados nas análises de associação. Pode-se verificar que, com qualquer dos conjuntos de dados (IC+MI, IC ou MI), as frequências genotípicas e alélicas geradas para cada um dos marcadores foram diversificadas, porém, quando consideradas as frequências para um mesmo marcador em cada arquivo, verificou-se que estas se mantiveram semelhantes, o que permitiu efetuar comparações dos resultados obtidos nas análises com cada um dos arquivos.
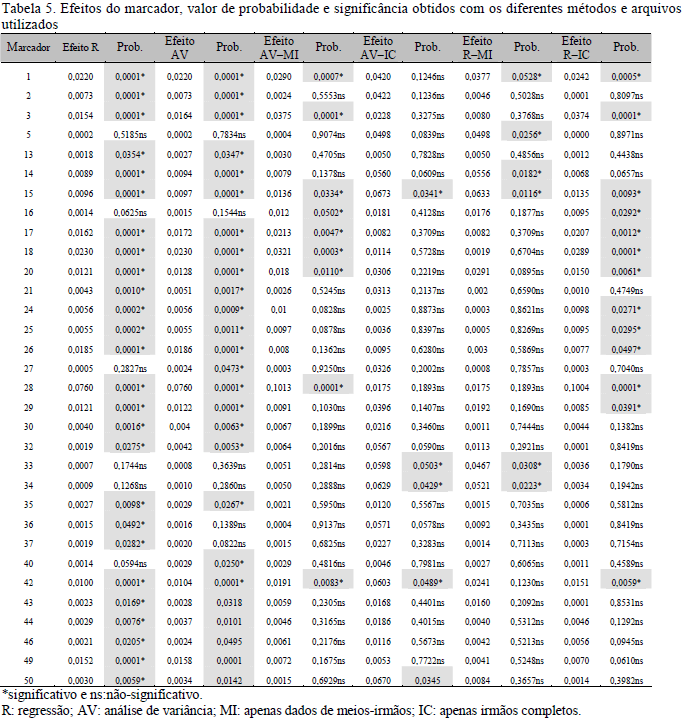
Na Tab. 5 são apresentados os resultados das análises de associação para todos os marcadores, ressaltando-se aqueles cujos efeitos foram significativos. Os resultados para M39 foram ignorados em função da existência de apenas dois dos três genótipos possíveis (Tab. 1). Considerando-se o arquivo IC+MI, como dado discreto (R) ou contínuo (AV), foi verificado que a análise de regressão de cada marcador sobre a variação genética não foi capaz de detectar associações entre o marcador e o QTL quando a magnitude dos efeitos do QTL foi pequena (<0,0015, para R, e <0,0020, para AV), nem mesmo assumindo-se valores menos conservadores de probabilidade (P<0,05) para a aceitação de associação. A magnitude mínima dos efeitos estimados, que permitiu a detecção de associação para cada valor de probabilidade, foi de 0,0015 (P<0,05), 0,0027 (P<0,01) e 0,0073 (P<0,0001).
Quando a magnitude dos efeitos foi elevada, mesmo sob frequências alélicas extremas, a possibilidade de detecção foi maior. Isso pode ser constatado para M01, M18 e M28 (P<0,0001). Para M02, M14 e M24, encontrou-se significância até mesmo para os efeitos de pequena magnitude, provavelmente porque as frequências alélicas estavam próximas às intermediárias. O efeito do marcador e a frequência de seus alelos são aspectos importantes na definição dos marcadores a serem utilizados para o mapeamento fino de regiões candidatas (Lynch e Walsh, 1998). Israel e Weller (2002) encontraram pequenas diferenças na estimativa dos efeitos do QTL ao utilizar diferentes métodos de detecção de QTL em dados em que pequena fração da população foi genotipada, assumindo-se diferentes frequências alélicas para os marcadores. Os autores, porém, não verificaram diferenças expressivas no vício das estimativas ou nas estimativas das médias dos errospadrão a frequências intermediárias ou extremas, apesar de os melhores resultados terem sido obtidos quando a frequência era intermediária.
Estes resultados são de grande interesse à validação de QTL identificados em algumas populações, com o objetivo de auxílio à seleção em outras populações. Uma vez comprovada a eficiência desta metodologia, a detecção/validação de associação poderá se dar em populações como a de um núcleo MOET; pequenas, mas com estrutura de família complexa o suficiente para esses estudos. Os resultados obtidos, embora promissores, devem, porém, ser avaliados com cautela, visto que as covariâncias entre irmãos completos (½) e meiosirmãos (¼) não foram consideradas no modelo e podem, portanto, viciar os resultados. Métodos que consideram a covariância genética entre parentes, neste caso entre irmãos completos e meios-irmãos, deverão ser implementados para confirmação dos resultados encontrados neste estudo.
Ao comparar os resultados das regressões, utilizando-se o arquivo IC+MI, quando a variável foi considerada classificatória (R) àqueles em que a variável foi considerada contínua (AV), verificaram-se pequenas diferenças na significância dos efeitos, sendo que M36 e M37 foram significativos (P<0,05 e P<0,03, respectivamente) apenas para R, enquanto M27 e M40 (P<0,05 e P<0,02, respectivamente), apenas para AV (Tab. 5). Nas análises comparativas dos resultados de associação obtidos por meio dos diferentes métodos (R e AV) e apresentados nas Tab. 6 e 7, observou-se alta correlação entre os resultados para o efeito do marcador (r=0,99) e sua significância (r=0,88), considerando-se todos os resultados. Se apenas valores mais conservadores de probabilidade (P<0,01) fossem considerados nas análises de correlação, esta se tornaria igual a um, mostrando que tais métodos teriam a mesma eficiência para detectar a associação entre marcador e o suposto QTL com dados de IC+MI. Segundo Haley e Knott (1992), as análises de regressão simples para detecção de QTL poderão ser mais eficientes se utilizarem marcadores flanqueadores.
Os resultados das análises dos diferentes conjuntos de dados e métodos mostraram também que as AV, utilizando apenas informações de MI, e as R, utilizando apenas informações de IC, apresentaram alta correlação com AV (IC+MI) e R(IC+MI), e detectaram valores semelhantes para os efeitos dos marcadores. Porém, considerando-se os valores de probabilidade, essas análises não detectaram significância na associação do marcador ao suposto QTL. Tais resultados podem ser atribuídos ao número de dados destes arquivos, pois, neste caso, a acurácia das estimativas da variância genética será menor, diminuindo o poder de detecção de alguns QTL. Apesar da diminuição significativa do poder de detecção de associação, uma vez que não se conhecem a fase de ligação e o grau de desequilíbrio de ligação e que o número de indivíduos genotipados é pequeno, este estudo verificou que a utilização conjunta das informações IC+MI, por meio de R ou de AV, em populações com a estrutura de núcleos MOET, permitiu detectar a associação marcador-QTL (Le Roy et al., 1998).
Na Tab. 8 são apresentados o valor de probabilidade e a magnitude dos efeitos associados aos marcadores. Os marcadores M01, M03, M17, M18 e M28 foram os de maior efeito médio de substituição gênica (P<0,0001). Assumindo-se níveis menos conservadores de significância, apenas os marcadores M27 e M40 apresentaram desvio significativo (P<0,03 e P<0,05, respectivamente) de dominância. Para M32, os efeitos aditivo e de dominância foram significativos (P<0,02 e P<0,05, respectivamente), o que pode ser reflexo da interferência de um QTL próximo, fato que, em dados reais, poderia ser explicado também pela ação epistática. Porém, níveis menos conservadores de significância foram assumidos e, portanto, estes resultados devem ser avaliados com reserva.
CONCLUSÕES
Apesar da pouca sensibilidade dos delineamentos baseados em populações exogâmicas e de tamanho pequeno, a metodologia de análise conjunta de dados de famílias de irmãos completos e de meios-irmãos por meio de regressão permitiu detectar a associação entre o marcador e o suposto QTL, até mesmo em níveis de significância conservadores. A frequência alélica e o efeito do marcador foram aspectos importantes para a eficiência dessa metodologia.
AGRADECIMENTOS
Ao Prof. Ricardo Frederico Euclydes, do Departamento de Zootecnia da UFV, pela gentileza de gerar os dados utilizados neste estudo.
Recebido em 11 de setembro de 2008
Aceito em 15 de maio de 2009
Apoio: FAPEMIG
E-mail: gaby@cnpgl.embrapa.br
- BECKMAN, J.S.; SOLLER, M. Detection of linkage between marker loci and loci affecting quantitative traits in crosses between segregating populations. Theor. Appl. Genet., v.76, p.228-236, 1989.
- BINK M.C., JANSS L.L., QUAAS R.L. Markov chain Monte Carlo for mapping a quantitative trait locus in outbred populations. Genet Res, 75:231-241, 2000.
- BINK, M.C.; VAN ARENDONK, J.A. Detection of quantitative trait loci in outbred populations with incomplete marker data. Genetics, v.151, p.409-420, 1999.
- BRASCAMP, E.W.; VAN ARENDONK, J.A.M.; GROEN, A.F. Economic appraisal of the utilization of genetic markers in dairy cattle breeding. J. Dairy Sci, v.76, p.1204-1213, 1993.
- COPPIETERS, W.; KVASZ, A.; ARRANZ J.J. et al. The great-grand-daughter design: a simple strategy to increase the power of a grand-daughter design for QTL mapping. Genet. Res., v.74, p.189-199, 1999.
- DARVASI A., WEINREB A., MINKE V. et al. Detecting marker-QTL linkage and estimating QTL gene effect and map location using a saturated genetic map. Genetics, v.134, p.943-951,1993
- DEKKERS, J.C.M. Commercial application of marker-and gene-assisted selection in livestock: Strategies and lessons. J. Anim. Sci, v.82, p.313-328, 2004.
- EUCLYDES, R.F. Uso do sistema para simulação Genesys na avaliação de métodos de seleção clássicos e associados a marcadores moleculares. 1996. 149f. Tese (Doutorado) - Universidade Federal de Viçosa, Viçosa, MG.
- FERNANDO, R.L.; GROSSMAN, M. Marker assisted selection using best linear unbiased prediction. Genet. Sel. Evol., v.21, p.467-477, 1989.
- HALEY, C.S.; KNOTT, S.A. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity, v.69, p.315-324, 1992.
- ISRAEL, C.; WELLER, J.I. Estimation of quantitative trait loci effects in dairy cattle populations. J. Dairy Sci, v.85, p.1285-1297, 2002.
- JOKINEN, J.; VANHAMÄKI, T.; TAMMIRANTA, N. et al. Five years of ASMO breeding scheme. Jokioinen: MTT Agrifood Res. Finland, NJF, 2003. Disponível em: <http://portal.mtt.fi/portal/page/portal/AGRONET/YHTEISET_HANKEET/NJF/NJF2003/3.PDF>, Acessado em: 17 set. 2007.
- KASHI, Y.; HALLERMAN, E.; SOLLER, M. Marker-assisted selection of candidate bulls for progeny testing programmes. Anim. Prod, v.51, p.63-74, 1990.
- LE ROY, P.; ELSEN, J.M.; BOICHARD, D. et al. An algorithm for QTL detection in mixture of full and half sib families. In: WORLD CONGRESS ON GENETIC APPLIED TO LIVESTOCK PRODUCTION, 6., 1998, Armidale. Proceedings.. Armidale: WCGALP, 1998.
- LYNCH, M.; WALSH, B. Genetic and analysis of quantitative traits Sunderland: Sinauer Association, 1998. 980p.
- MARTINEZ, M.L.; VUKASINOVIC, N. Algoritmo para cálculo da proporção de genes idênticos por descendência para mapear QTL em famílias de meio-irmãs. Rev. Bras. Zootec, v.29, p.443-451, 2000.
- NICHOLAS, F.W. Discovery, validation and delivery of DNA markers. Aust. J. Exp. Agric, v.46, p.155-158, 2006.
- PENNA, V.M.; MADALENA, F.E.; ALVIM, M.T.T. Open MOET nucleus of selection in Guzerá. In: WORLD CONGRESS ON GENETIC APPLIED TO LIVESTOCK PRODUCTION, 6., 1998, Armidale. Proceedings... Armidale: WCGALP, 1998. (CD-ROM).
- SIMPSON, S.P. Detection of linkage between quantitative trait loci and restriction fragment length polymorphisms using inbred lines. Theor. Appl. Genet, v.77, p.815-819, 1989.
- STRATHIE, R.J.; McGUIRK, B.J. Developments with MOET dairy breeding scheme. Br. Cattle Breed. Club Dig., v.50, p.9-15, 1995.
- van der BEEK, S.; VAN ARENDONK, J.A.M.; GRÖEN, A.F. Power of two and three-generation QTL mapping experiments in an outbred population containing full-sib or half-sib families. Theor. Appl. Genet, v.91, p.1115-1124, 1995.
Datas de Publicação
-
Publicação nesta coleção
27 Ago 2009 -
Data do Fascículo
Ago 2009
Histórico
-
Aceito
15 Maio 2009 -
Recebido
11 Set 2008