Abstracts
PURPOSE: To determine podocyte number and GBM thickness in diabetic rats either under glycemic control or without glycemic control at 6 and 12 months after diabetes induction. METHODS: 100 wistar rats weighing 200-300g were divided into 6 groups: Normal group (N6 and N12- 25 rats); Diabetic group (D6 and D12- 25 rats), diabetic treated group ( DT 6 and DT 12- 25 rats) on insulin 1,8- 3,0 IU/Kg associated with acarbose (50mg to 100g of food) daily mixed in chow. Alloxan was injected intravenously in a dose of 42 mg/Kg of weight. Body weight, waterintake, 24-h diuresis, glycemia and glucosuria were determined before induction, 7 and 14 days after induction and monthly thereafter. Treatment started at day 14. Three groups were sacrificed at 6 months (N6,D6, DT6) and 3 groups at 12 months (N12, D12, DT12) with the renal tissue being prepared for electron microscopy. RESULTS: Glycemia in DT6¨and in DT12 was significantly different from that in D6 and D12 rats and similar to that in N6 and N12 animals. The number of podocytes in DT6 was not different from that in N6 and D6 (median = 11); the number of podocytes in DT12 (median = 11) differed from that in D12 (median = 8), but not from that in N12 (median = 11). GBM thickness in D6 (0.18 micrometers) was lower than in D12 (0.29 micrometers); while in DT6 (0.16 micrometers) it was lower than in D6 (0.18 micrometers). In DT12 (0.26 micrometers), it was lower than in D12 (0.29 micrometers). CONCLUSION: The control of hyperglycemia prevented GBM thickening in early and late (12 mo) alloxan diabetic nephropathy and podocyte number reduction.
Diabetes Mellitus; Experimental; Microscopy; Electron; Podocytes; Rats
OBJETIVO: Avaliar o número de podócitos e espessamento da membrana basal glomerular (MBG) em ratos diabéticos com e sem controle glicêmico com 6 e 12 meses da indução. MÉTODOS: 100 ratos Wistar com 200-300g compuseram 6 grupos: Normal (N6, N12 - 25 animais) Diabético (D6,D12 - 25 animais) e diabético tratado com insulina 1,8 a 3,0 U/Kg e acarbose misturada a ração (50g para cada 100g de ração) (DT6 e DT12 - 25 animais). Aloxana foi ministrada via endovenosa na dose de 42mg/Kg. Peso, ingestão hídrica e diurese de 24 horas e glicemia e glicosúria foram determinados antes da inoculação, 7 e 14 dias após e mensalmente. No 14ª dia foi iniciado o tratamento. Três grupos de animais (N6, D6 e DT6) foram sacrificados no 6° mês e três grupos (N12, D12 e DT12), no 12ª mês sendo o tecido renal processado para estudo à microscopia eletrônica. RESULTADOS: A glicemia dos animais DT6 e DT12 diferiram significativamente, dos ratos D6 e D12, e não diferiram dos grupos N6 e N12. O número de podócitos do grupo DT6 não diferiu de N6 e D6 (mediana=11); o número de podócitos de DT12 (mediana=11) diferiu de D12 (mediana=8) e não diferiu de N12 (mediana=11). O espessamento da MBG de D6 (0,18 micrômetros) foi menor que D12 (0,29 micrômetros); de DT6 (0,16 micrômetros) foi menor que D6 (0,18 micrômetros) e de DT12 (0,26 micrômetros) foi menor que D12 (0,29 micrômetros). CONCLUSÃO: O controle da hiperglicemia preveniu o espessamento da MBG na nefropatia diabética aloxânica precoce (6 meses) e tardia (12 meses), e a diminuição do número de podócitos.
Diabetes Mellitus Experimental; Microscopia Eletrônica; Podócitos; Ratos
ORIGINAL ARTICLE
DIABETES MELLITUS, EXPERIMENTAL
Reduction of podocytes number in late diabetic alloxan nephropathy. Prevention by glycemic control1 1 Research performed at Experimental Laboratory Technique of Surgery, Department of Surgery and Orthopedics, School of Medicine, São Paulo State University (UNESP), Botucatu, Brazil.
Redução do número de podócitos na fase tardia da nefropatia diabética aloxânica. Prevenção pelo controle glicêmico
Célia Sperandéo MacedoI; Mauro Masson LercoII; Sônia Maria CapellettiIII; Reinaldo José SilvaIV; Daniela de Oliveira PinheiroV; César Tadeu SpadellaVI
IMD, PhD, Associate Professor, Department of Pediatrics, School of Medicine, UNESP, Botucatu, Brazil
IIMD, PhD, Assistant Professor, Department of Surgery and Orthopedics, School of Medicine, UNESP, Botucatu, Brazil
IIIBiologist, Experimental Laboratory Technique of Surgery, Department of Surgery and Orthopedics, School of Medicine, UNESP, Botucatu, Brazil
IVBiologist, MD Fellow, Department of Morphology, Institute of Biosciences, UNESP, Botucatu, Brazil
VBiologist, PhD, Assistant Professor, Department of Parasitology, Institute of Biosciences, UNESP, Botucatu, Brazil
VIMD, PhD, Associate Professor, Department of Surgery and Orthopedics. School of Medicine, UNESP, Botucatu, Brazil
Correspondence Correspondence: Dr. César Tadeu Spadella UNESP Depto de Cirurgia e Ortopedia Rubião Junior, s/n 18618-970 Botucatu São Paulo Brasil spadella@fmb.unesp.br
ABSTRACT
PURPOSE: To determine podocyte number and GBM thickness in diabetic rats either under glycemic control or without glycemic control at 6 and 12 months after diabetes induction.
METHODS: 100 wistar rats weighing 200-300g were divided into 6 groups: Normal group (N6 and N12- 25 rats); Diabetic group (D6 and D12- 25 rats), diabetic treated group ( DT 6 and DT 12- 25 rats) on insulin 1,8- 3,0 IU/Kg associated with acarbose (50mg to 100g of food) daily mixed in chow. Alloxan was injected intravenously in a dose of 42 mg/Kg of weight. Body weight, waterintake, 24-h diuresis, glycemia and glucosuria were determined before induction, 7 and 14 days after induction and monthly thereafter. Treatment started at day 14. Three groups were sacrificed at 6 months (N6,D6, DT6) and 3 groups at 12 months (N12, D12, DT12) with the renal tissue being prepared for electron microscopy.
RESULTS: Glycemia in DT6¨and in DT12 was significantly different from that in D6 and D12 rats and similar to that in N6 and N12 animals. The number of podocytes in DT6 was not different from that in N6 and D6 (median = 11); the number of podocytes in DT12 (median = 11) differed from that in D12 (median = 8), but not from that in N12 (median = 11). GBM thickness in D6 (0.18 micrometers) was lower than in D12 (0.29 micrometers); while in DT6 (0.16 micrometers) it was lower than in D6 (0.18 micrometers). In DT12 (0.26 micrometers), it was lower than in D12 (0.29 micrometers).
CONCLUSION: The control of hyperglycemia prevented GBM thickening in early and late (12 mo) alloxan diabetic nephropathy and podocyte number reduction.
Key words: Diabetes Mellitus, Experimental. Microscopy, Electron. Podocytes. Rats.
RESUMO
OBJETIVO: Avaliar o número de podócitos e espessamento da membrana basal glomerular (MBG) em ratos diabéticos com e sem controle glicêmico com 6 e 12 meses da indução.
MÉTODOS: 100 ratos Wistar com 200-300g compuseram 6 grupos: Normal (N6, N12 25 animais) Diabético (D6,D12 25 animais) e diabético tratado com insulina 1,8 a 3,0 U/Kg e acarbose misturada a ração (50g para cada 100g de ração) (DT6 e DT12 25 animais). Aloxana foi ministrada via endovenosa na dose de 42mg/Kg. Peso, ingestão hídrica e diurese de 24 horas e glicemia e glicosúria foram determinados antes da inoculação, 7 e 14 dias após e mensalmente. No 14ª dia foi iniciado o tratamento. Três grupos de animais (N6, D6 e DT6) foram sacrificados no 6° mês e três grupos (N12, D12 e DT12), no 12ª mês sendo o tecido renal processado para estudo à microscopia eletrônica.
RESULTADOS: A glicemia dos animais DT6 e DT12 diferiram significativamente, dos ratos D6 e D12, e não diferiram dos grupos N6 e N12. O número de podócitos do grupo DT6 não diferiu de N6 e D6 (mediana=11); o número de podócitos de DT12 (mediana=11) diferiu de D12 (mediana=8) e não diferiu de N12 (mediana=11). O espessamento da MBG de D6 (0,18 micrômetros) foi menor que D12 (0,29 micrômetros); de DT6 (0,16 micrômetros) foi menor que D6 (0,18 micrômetros) e de DT12 (0,26 micrômetros) foi menor que D12 (0,29 micrômetros).
CONCLUSÃO: O controle da hiperglicemia preveniu o espessamento da MBG na nefropatia diabética aloxânica precoce (6 meses) e tardia (12 meses), e a diminuição do número de podócitos.
Descritores: Diabetes Mellitus Experimental. Microscopia Eletrônica. Podócitos. Ratos.
Introduction
The importance of podocyte lesions in diabetic nephropathy has been established in patients with Type 2 diabetes. Podocyte loss in early progressive glomerulosclerosis has been described in North-American PIMA Indians1,2, and in patients with Type 1 diabetes3.Reduced podocyte number has been seen after 6 months in rats with streptozotocin (STZ)-induced diabetes, compared with normal rats4,5. In alloxan diabetic rats an evident podocyte reduction has been observed concomitantly with an increased excretion of protein in urine 12 months after induction6. There is an inverse relationship between glucose homeostasis an renal damage7,8. Maintaining glycemia levels close to normal is seldom achieved by conventional treatment. One of the reasons is that carbohydrate absorption in the intestinal tract does not occur at the same rate and speed as insulin absorption from the injection site9. Acarbose inhibits glucosidase, an enzyme that hydrolyzes carbohydrates in the intestine10,11, and decreases postprandial glycemia in diabetic patients treated with insulin12. Diabetic animals treated with acarbose combined with insulin have been reported to develop lower glomerular basement membrane (GBM) thickeningthat was clearly identifiable after six months of diabetes13.The increasing interest in the investigation of podocytes in diabetic nephropathy motivated this experimental study. Our objective was to evaluate whether the early treatment of alloxan diabetes with insulin combined with acarbose for 6 and 12 months results in the preservation of podocyte number and GBM thickening.
Methods
One hundred male and female Wistar rats, approximately three months old and weighing 200-300g, were divided into six experimental groups: Normal Group (N6 and N12), consisting of 25 healthy animals; Diabetic Group (D6 and D12), including 25 rats with alloxan-induced diabetes (42 mg/Kg); and Diabetic Treated Group (DT6 and DT12), comprising 25 rats with alloxan-induced diabetes treated with slow insulin (Monotard R, Novo Nordisk Farmacêutica do Brasil, Ltda, São Paulo, SP) associated with Acarbose (Bayer do Brasil, São Paulo). Insulin was administered at an initial dose of 1,8 to 30 IU/Kg which was adjusted according to daily glycosuria and ketonuria. Acarbose (50mg/100 g of food) was mixed in chow and offered to the animals on a daily basis. Only the diabetic rats with glycemia higher than 200mg%, glycosuria higher than 3000mg% and clear signs of severe diabetes 7 and 14 days after induction were included in the study. The parameters evaluatedincluded weight, urine volume,24-h water intake, fasting blood glucose levels (mg%), 24-h glycosuria (mg%). Treatment started at day 14 day and was maintained until sacrifice. After 6 months, 10 Normal rats (N6); 13 Diabetic rats (D6), and 8 Diabetic Treated rats (DT6) were sacrificed. After 12 months, 12 Normal rats (N12); 11 Diabetic rats (D12) and 6 Diabetic Treated rats (DT12) were sacrificed. This experiment was approved by the Animal Research Ethics Committee of Botucatu Medical School São Paulo State University/UNESP.
Morphometric study
The right kidney of four animals from each group was removed, cut into 3-4 mm cylinders and fixed in 1% glutaraldehyde in phosphate buffer, postfixed in 2% osmium sulfate in phosphate buffer, ph 7,4, for 2 h (4ºC), dehydrated in alcohol, and embedded in epon resin. Blocks were prepared by manual trimming. Semithin sections were prepared for light microscopy and ultrathin sections were stained with lead citrate and examined under electron microscopy. Photomicrographs of 5 blocks per rat (6 photomicrographs per block) were taken until a minimum of 100 slits per animal was obtained at 42,000 X final magnification. Podocyte slits with detectable linear images of the slit diaphragm were photographed and counted. The pores where the sectioning angle made the evaluation of the slit diaphragm impossible were ignored. A total of 115 photomicrographs of the normal group (N6 and N12), 138 of the diabetic group (D6 and D12) and 140 of the diabetic treated group (DT6 and DT12) were taken.Subsequently, the computer image analyzer system "Qwin Lite 2,5-Leica" was used to determinepodocyte numbe,r slit diaphragm number and GBM thickness. All measurements were performed by the same technician, who was unaware of the experimental group to which the material belonged. Data were analysed by Mann-Whitney test. The software sigma-stat 2.0 was used in the comparison of data.
Results
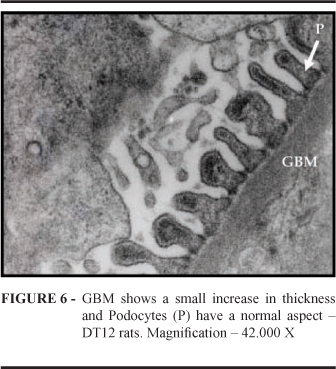
Blood glucose levels are shown in Figure 1. No difference between Normal and Diabetic Treated animals was observed(p< 0,001). The number of podocytes (Figure 2) in Normal animals did not differ from that in Diabetic and Diabetic Treated animals at 6 months . At 12 months, however, podocyte number in the Diabetic treated group differed from that in the Diabetic Group, but showed no difference when compared with the Normal group (p< 0,001). Data regarding GBM thickening are presented in Figure 3. In Diabetic animals, GBM thickness showed a statistical difference, i.e., it was lower at 6 months than at 12 months. Nonetheless, in the Diabetic Treated Group, GBM thickness was lower than in the Diabetic Group, both at 6 months and at 12 months (p< 0,001). GBM thickness and podocyte aspect 12 months after diabetes induction in normal, Diabetic and Diabetic Treated rats were compared (Figures 4, 5 and 6). In Diabetic Treated animals, GBM thickness was lower than in diabetic rats and podocyte number was similar to that in normal animals.
Discussion
Podocytes or visceral epithelial cells are highly differentiated glomerular cells which cover GBM. They areunable to replicate, form the filtration slit structure that prevents protein to escape into urine14,15. Considerable evidence in experimental animals indicates that podocyte disruption contributes to the development of glomerular sclerosis16,17,18. An increase in glomerular pressure causes mechanical stress in the glomerular cells podocytes among them. Cyclic mechanical stretch cultures induces hypertrophy in cultured mouse podocytes19. In diabetic nephropathy, due to glomerular hypertrophy and hypertension, podocytes are damaged and lost in urine20,21 producing bare GBM areas with consequent sinechiae formation between GBM and Bowman's capsule and the development of glomerular sclerosis22. Podocyte number reduction in STZ diabetic rats after 24 weeks as demonstrated by counting the number of slit pores per unit lengh of the glomerular basement by electron microscopy4 or by using immunohistochemical techniques5 has been recently described. In these studies, the experimental groups were treated with agents that decrease glomerular hypertension, and glycemic levels were maintained. Thus podocyte number reduction was attenuated4,5. The role of glycemic control in renal lesions was investigated herein. In a previous work, we used electron microscopy to study alloxan diabetic rats for 48 weeks, and demonstrated the presence of significant proteinuria, reduction in podocyte number, and increased GMB thickness6. In this study, glycemic control attenuated glomerular changes; GBM thickness was not so intense 6 and 12 months after diabetes induction and the podocyte number at 12 months was similar to that in normal animals when continuously using insulin combined with an oral antihyperglycemiant. These results were not observed in our previous study using insulin alone for 12 months23. With regard to GBM, it was thicker at 12 months than at 6 months in the three groups. Glycemic control clearly attenuated this type of lesion. These findings are similar to others reported in the literature7,13. Glycemia differed between treated diabetic rats and diabetic rats. However, the number of podocytes at 6 months did not differ among the three groups. Immuno histochemical techniques would be worthwhile in confirming these results5,24,25. Glycemic control probably prevented glomerular hypertension and podocyte injury preserving podocyte number. In diabetes, GBM synthesis is rapid and degradation is low . Many mediators have already been identified, among them is growth factor beta that stimulates the glomerular collagen matrix, including GBM, and inhibits enzymes (proteases) that degrade collagen28,29,30. It is possible that glycemic control avoided these mechanisms. Electron microscopy 6 and12 months after diabetes induction provides consistent results that reflect the preservation of podocyte number and the attenuation of GBM thickening under relatively tight glycemic control.
Conclusion
That glycemic control prevented podocyte damage and the GBM thickening , two basic elements of experimental diabetic nephropathy. It is important to emphasize that no study describing podocyte changes in rats under glycemic control for this long is currently available in the literature.
Received: March 14, 2007
Review: May 15, 2007
Accepted: June 14, 2007
Conflict of interest: none
Financial source: none
- 1. Lemley KV, Blouch K, Abdullah I, Boothroyd DB, Bennett PH, Myers BD, Nelson RG. Glomerular perm selectivity at the onset of nephropathy in type 2 diabetes mellitus J Am Soc Nephrol 2000;11:2095-105.
- 2. Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia. 1999;42:1341-4.
- 3. Steffes MW, Schmidt D, McCrery R. Glomerular cell number in normal subjects and in type I diabetic patients. Kidney Int. 2001;59:2104-13.
- 4. Mifsud SA, Allen TJ, Bertam JF, Hulthen UL, Kelly DJ, Cooper ME, Wilkinson-Berka JL, Gilbert RE. Podocyte foot process boadening in experimental diabetic nephropathy: amelioration with renin-angiotensin blockade. Diabetologia. 2001;44:878-82.
- 5. Gross ML, Shakmak A, El, Szabo A, Koch A, Kuhlmann A, Munter K, Ritz E, Amann K. ACE inhibitors but not endothelin receptor blockers prevent podocyte loss in early diabetic nephropathy. Diabetologia. 2003;46:856-68.
- 6. Lerco MM, Macedo CS, Silva RJ, Pinheiro DO, Spadella CT. The number of podocyte and slit diaphragm is decreased in experimental diabetic nephropathy. Acta Cir Bras. 2006;21(2):87-91.
- 7. Rash R. Prevention of diabetic glomerulopathy in streptozotocin diabetic rats by insulin treatment: glomerular basement membrane thickness. Diabetologia. 1979;16:319-24.
- 8. Rash R. Prevention of diabetic glomerulopathy in streptozotocin diabetic rats by insulin treatment: the mesangial regions. Diabetologia. 1979;17:243-8.
- 9. Job D, Esehwege E, Guyot G, Aubry JP and Tchobroutsky G. Effect of multiple daily insulin injections on the course of diabetic retinopathy. Diabetes. 1976;25:463-9.
- 10. Schmidt DD, Frommer W, Jenge B, Muller L, Wingender W, Truscheit E, Shafer D. Alpha-glucosidase inhibitors: new complex oligosaccharides of microbial origin. Naturwissenschaften. 1977;64:635-6.
- 11. Puls W, Keup U, Krausse HP, Thomas G and Hoffmeister F. Glucosidase inhibition, a new approach to the treatment of diabetes, obesity and hyperlipoproteinemia. Naturwissenschaften. 1977;64:536-7.
- 12. Sachse G and Willms B. Effect of the alpha-glucosidase inhibitor Bay-g-5421 on blood glucose control of sulphonylurea treated diabetics and insulin treated diabetics. Diabetologia. 1979;17:287-90.
- 13. Macedo CS, Silva MD, Spadella CT, Breim LC, Capeletti S, Mercadante MCS, Hernandes D, Macedo AR. Effect of long-term treatment with insulin and/or acarbose on glomerular basement membrane thickening in alloxan-diabetic rats. Braz J Med Biol Res. 1996;29:1329-35.
- 14. Kriz W. Progressive renal failure: inability of podocytes to replicate and the consequences for development of glomeruloscleroses. Nephrol Dial Transplant. 1996;11:1738-42.
- 15. Kerjaschki D. Dysfunction of cell biological mechanisms of visceral epithelial cell (podocytes) in glomerular diseases. Kidney Int. 1994;45:300-13.
- 16. Kriz W, Elger M, Nagata M. The role of podocytes in the development of glomerular sclerosis. Kidney Int. 1994;45(42):S64-72.
- 17. Kriz W, Gretz N, Lemley KV. Progression of glomerular disease: is the podocyte the culprit ? Kidney Int. 1998;54:687-97.
- 18. Adler S. Diabetic nephropathy: linking histology, cell biology and genetics. Kidney Int. 2004;66:2095-106.
- 19. Petermann AT, Pippin J, Durvasula R, Pichler R, Hikomura K, Monkawa T, Couser WG and Shankland SJ. Mechanical stretch induces podocyte hypertrophy in vitro. Kidney Int. 2005;67:157-66.
- 20. Nakamura T, Ushiwama C, Suzuki S, Hara M, Shimada N, Ebihara I, Koide H. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant. 2000;15:1379-83.
- 21. Viera Jr JM, Fujihara CK, Zatz R. Mecanismos de progressão da doença renal. In: Toporovski J, Mello VR, Mrtini Filho D, Benini V, Andrade OVB. Nefrologia pediátrica. 2ed. Rio de Janeiro: Guanabara Koogan; 2006. p.67-76.
- 22. Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13:3005-15.
- 23. Spadella CT, Lerco MM, Machado JLM, Macedo CS. Long-term effects of insulin therapy, islet transplantation and pâncreas transplantation in the prevention of glomerular changes in kidneys of alloxan-induced diabetic rats. Transplant Proc. 2005;37:3468-71.
- 24. Hoffmann S; Podlich D; Hahnel B; Kriz W; Gretz N. Angiotensin II Type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol. 2004;15:1475-87.
- 25. Attia DM; Feron O; Goldschmeding R; Radermakers LH; Vaziri ND; Boer P; Balligand JL;Koomans HA; Joles JA. Hypercholesterolemia in rats induces podocytes stresss and decreases renal cortical nitric oxide synthesis via an angiotensin II type I receptor-sensitive mechanism. J Am Soc Nephrol. 2004;15:949-57.
- 26. Hostetter TH; Troy JL; Brenner BM. Glomerular hemodynamics in experimental diabetes mellitus. Kidney Int. 1981;19:410-5.
- 27. Border WA; Yamamoto T; Noble NA. Transforming growth factor in diabetic nephropathy. Diabetes Metabol Rev. 1996;12:309-39.
- 28. Reckelhoff JF; Tygart VL; Mitias MM; Walcott JL. Stz-induced diabetes results in decreased activity of glomerular cathepsin and metalloprotease in rats. Diabetes. 1993;42:1425-32.
- 29. Sharma K; Ziyadeh FN. Biochemical events and cytokine interactions linking glucose metabolism to the development of diabetic nephropaty. Semin Nephrol. 1997;17:80-92.
- 30. Ziyadeh FN; Sharma K. Role of transforming growth factor-beta in diabetic glomerulosclerosis and renal hypertrophy. Kidney Int. 1995;51:534-6.
Publication Dates
-
Publication in this collection
27 Sept 2007 -
Date of issue
Oct 2007
History
-
Reviewed
15 May 2007 -
Received
14 Mar 2007 -
Accepted
14 June 2007