Abstracts
PURPOSE: Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that displays a rapid evolution. Current treatments have failed to revert clinical symptoms because the mechanisms involved in the death of motoneuron are still unknown. Recent publications have put non-neuronal cells, particularly, astrocyte and microglia, in the scenario of pathophisiology of the disease. Animal models for ALS, particularly transgenic mice expressing the human SOD1 gene with a G93A mutation (hSOD1), are available and display the phenotype of the disease at cellular and clinical levels. However, it is a lack of detailed information regarding the methods to study the disease in vitro to better understand the contribution of non-neuronal cells in the onset and progression of the pathology. METHODS: Colonies of Swiss mice and transgenic mice expressing hSOD1 mutation as well as non-transgenic controls (wild-type) were amplified after a genotyping evaluation. Disease progression was followed behaviorally and mortality was registered. Highly purified primary cultures of astrocytes and microglia from mouse spinal cord were obtained. Cells were identified by means of GFAP and CD11B immunocytochemistry. The purity of astroglial and microglial cell cultures was also accompanied by means of Western blot and RT-PCR analyses employing a number of markers. RESULTS: The disease onset was about 105 days and the majority of transgenic mice displayed the disease symptoms by 125 days of age and reached the endpoint 20 days later. A substantial motor weakens was registered in the transgenic mice compared to wild-type at the end point. Immunocytochemical, biochemical and RT-PCR analyses demonstrated a highly purified primary cultures of spinal cord astrocytes and microglia. CONCLUSION: It is possible to achieve highly purified primary cultures of spinal cord astrocytes and microglia to be employed in cellular and molecular analyses of the influence of such non-neuronal cells in the pathophysiology of ALS.
Amyotrophic Lateral Sclerosis; Astrocytes; Microglia; Neurons; Cell Culture Techniques; PCR; Mice; Transgenic
OBJETIVO: A esclerose lateral amiotrófica (ELA) é uma doença neurodegenerativa fatal com evolução rápida. Os tratamentos atualmente disponíveis falham em reverter os sintomas porque os mecanismos envolvidos na morte do neurônio motor ainda não são conhecidos. Publicações recentes colocam as células não neuronais, particularmente o astrócito e a microglia, no cenário da fisiopatologia da doença. Modelos animais para a ELA, particularmente os camundongos transgênicos que expressam o gene da SOD1 humana (hSOD1) mutante estão disponíveis e mostram o fenótipo da doença ao nível celular e clínico. Entretanto, informações detalhadas são escassas sobre os métodos de estudo da doença in vitro para a melhor compreensão da participação das células não neuronais no início e na progressão da patologia. MÉTODOS: Colônias de camundongos Swiss e camundongos transgênicos que expressam a hSOD1 mutante assim como os controles não transgênicos (selvagem) foram amplificadas após avaliação genotípica. A progressão da doença foi acompanhada pelo comportamento e a mortalidade foi registrada. Culturas primárias altamente purificadas de astrócitos e microglia da medula espinal dos camundongos foram obtidas. As células foram identificadas pela immunocitoquímica da GFAP e CD11B. A pureza das culturas de astrócitos e microglia foi acompanhada pelas análises do Western blot e RT-PCR empregando-se marcadores específicos. RESULTADOS: Os primeiros sinais da doença ocorreram por volta dos 105 dias de vida e a maioria dos camundongos transgênicos já estava com a doença manifestada aos 125 dias de idade e alcançaram o estágio terminal aproximadamente 20 dias depois. Fraqueza substancial da força muscular foi registrada nos animais transgênicos comparados com os animais selvagens. Análises imuncitoquímica, bioquímica e pelo RT-PCR demonstraram culturas primárias altamente purificadas de astrócito e microglia da medula espinal dos camundongos. CONCLUSÃO: É possível obter culturas purificadas de astrócitos e microglia da medula espinal do camundongo a ser empregadas em análises celulares e moleculares da influência destas células não neuronais na fisiopatologia da ELA.
Esclerose Amiotrófica Lateral; Astrócitos; Microglia; Neurônios; Técnicas de Cultura de Células; PCR; Camundongos Transgênicos
11 - ORIGINAL ARTICLE
MODELS, BIOLOGICAL
The review of the methods to obtain non-neuronal cells to study glial influence on Amyotrophic Lateral Sclerosis pathophysiology at molecular level in vitro1 1 Research performed at Neuroregeneration Center (LIM 45), Department of Neurology, School of Medicine, University of São Paulo (USP), Brazil.
Revisão dos métodos de obtenção de células não neuronais para o estudo da fisiopatologia da Esclerose Lateral Amiotrófica ao nível molecular in vitro
Juliana Milani ScorisaI; Tatiana DuoblesII; Gabriela Pintar de OliveiraII; Jessica Ruivo MaximinoIII; Gerson ChadiIV
IMaster in Sciences, Fellow of Department of Neurology, USP, São Paulo, Brazil
IIFellow PhD degree of Neurology, School of Medicine, USP, São Paulo, Brazil
IIIPhD, Researcher, Experimental Neurology, Department of Neurology, School of Medicine, USP, São Paulo, Brazil
IVPhD, Full Professor, Experimental Neurology, Department of Neurology, School of Medicine, USP, São Paulo, Brazil
Correspondence Correspondence: Gerson Chadi, M.D., Full Professor Department of Neurology University of São Paulo Av. Dr. Arnaldo, 455/2119 01246-903 São Paulo SP Brazil Phone: (55 11)3061-7460 gerchadi@usp.br
ABSTRACT
PURPOSE: Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that displays a rapid evolution. Current treatments have failed to revert clinical symptoms because the mechanisms involved in the death of motoneuron are still unknown. Recent publications have put non-neuronal cells, particularly, astrocyte and microglia, in the scenario of pathophisiology of the disease. Animal models for ALS, particularly transgenic mice expressing the human SOD1 gene with a G93A mutation (hSOD1), are available and display the phenotype of the disease at cellular and clinical levels. However, it is a lack of detailed information regarding the methods to study the disease in vitro to better understand the contribution of non-neuronal cells in the onset and progression of the pathology.
METHODS: Colonies of Swiss mice and transgenic mice expressing hSOD1 mutation as well as non-transgenic controls (wild-type) were amplified after a genotyping evaluation. Disease progression was followed behaviorally and mortality was registered. Highly purified primary cultures of astrocytes and microglia from mouse spinal cord were obtained. Cells were identified by means of GFAP and CD11B immunocytochemistry. The purity of astroglial and microglial cell cultures was also accompanied by means of Western blot and RT-PCR analyses employing a number of markers.
RESULTS: The disease onset was about 105 days and the majority of transgenic mice displayed the disease symptoms by 125 days of age and reached the endpoint 20 days later. A substantial motor weakens was registered in the transgenic mice compared to wild-type at the end point. Immunocytochemical, biochemical and RT-PCR analyses demonstrated a highly purified primary cultures of spinal cord astrocytes and microglia.
CONCLUSION: It is possible to achieve highly purified primary cultures of spinal cord astrocytes and microglia to be employed in cellular and molecular analyses of the influence of such non-neuronal cells in the pathophysiology of ALS.
Key words: Amyotrophic Lateral Sclerosis. Astrocytes. Microglia. Neurons. Cell Culture Techniques. PCR. Mice, Transgenic.
RESUMO
OBJETIVO: A esclerose lateral amiotrófica (ELA) é uma doença neurodegenerativa fatal com evolução rápida. Os tratamentos atualmente disponíveis falham em reverter os sintomas porque os mecanismos envolvidos na morte do neurônio motor ainda não são conhecidos. Publicações recentes colocam as células não neuronais, particularmente o astrócito e a microglia, no cenário da fisiopatologia da doença. Modelos animais para a ELA, particularmente os camundongos transgênicos que expressam o gene da SOD1 humana (hSOD1) mutante estão disponíveis e mostram o fenótipo da doença ao nível celular e clínico. Entretanto, informações detalhadas são escassas sobre os métodos de estudo da doença in vitro para a melhor compreensão da participação das células não neuronais no início e na progressão da patologia.
MÉTODOS: Colônias de camundongos Swiss e camundongos transgênicos que expressam a hSOD1 mutante assim como os controles não transgênicos (selvagem) foram amplificadas após avaliação genotípica. A progressão da doença foi acompanhada pelo comportamento e a mortalidade foi registrada. Culturas primárias altamente purificadas de astrócitos e microglia da medula espinal dos camundongos foram obtidas. As células foram identificadas pela immunocitoquímica da GFAP e CD11B. A pureza das culturas de astrócitos e microglia foi acompanhada pelas análises do Western blot e RT-PCR empregando-se marcadores específicos.
RESULTADOS: Os primeiros sinais da doença ocorreram por volta dos 105 dias de vida e a maioria dos camundongos transgênicos já estava com a doença manifestada aos 125 dias de idade e alcançaram o estágio terminal aproximadamente 20 dias depois. Fraqueza substancial da força muscular foi registrada nos animais transgênicos comparados com os animais selvagens. Análises imuncitoquímica, bioquímica e pelo RT-PCR demonstraram culturas primárias altamente purificadas de astrócito e microglia da medula espinal dos camundongos. CONCLUSÃO: É possível obter culturas purificadas de astrócitos e microglia da medula espinal do camundongo a ser empregadas em análises celulares e moleculares da influência destas células não neuronais na fisiopatologia da ELA.
Descritores: Esclerose Amiotrófica Lateral. Astrócitos. Microglia. Neurônios. Técnicas de Cultura de Células. PCR. Camundongos Transgênicos.
Introduction
Amyotrophic lateral sclerosis (ALS) is a human devastating and progressive neurodegenerative disorder that result in the selective death of motoneurons in the central nervous system. This progressive motoneuron degeneration culminates in muscle wasting and patient death on average three to five years after onset of the disease from respiratory failure1.
The main neuronal cells undertaking degeneration are the upper giant motoneurons of layer V of the motor cortex and their projecting lower motoneurons that are located in the motor nuclei of the brainstem and in the anterior horn of the spinal cord. The degeneration of the lower motoneurons that project to skeletal muscles is responsible for the majority of the neurological impairments of ALS patients. Affected motoneurons often contain characteristic inclusions in their perikarya, dendrites and axons however, more recently in the era of molecular biology, new mechanisms of ALS have been elucidated and the involvement of non-neuronal cells have been described2.
The knowledge on etiology and pathogenesis of ALS motoneuron death has been gained on the inherent limitation of human tissue from patients at the terminal stages, which makes it difficult to interpret events in the intermediate phases of the disease progression. Therefore, the search for animal models of ALS to gain insights into both the mechanisms of motoneuron death and therapeutic targets has allowed new information on the pathogenesis of ALS3. In fact, animal models have provided substantial advance into mechanisms that lead to motoneuron degeneration4.
The majority of ALS cases are apparently sporadic, with 90-95% of patients presenting disease symptoms without a familial history of the pathology, being the remaining 5-10% patients included as a familial ALS group, and about 25% of familial cases are caused by dominant mutations in the Cu/Zn super oxide dismutase (SOD1) gene5.
Identification of pathogenic alleles of SOD1 has led to the generation of transgenic mouse and rat, models for ALS studies. Overproduction of mutant human SOD1 protein in these models leads to a late-onset and a progressive motoneuron neurodegenerative disease. Such models have provided the identification of intrinsic pathogenic characteristics of ALS motoneurons, including the formation of protein aggregates, cytoskeletal abnormalities, proteosome dysfunction and increased sensitivity to cell death signals as well as the glial reactivity in the disease evolution6.
SOD1 mutant enzyme retains full enzymatic activity. Furthermore, experiments conducted in transgenic mice and rats bearing ALS-associated human SOD1 (hSOD1) mutations as well as SOD1 knockout mice indicate that SOD1-mediated toxicity is not due to a loss of function but rather to a toxic gain of function of the mutant protein. Taking together, results from in vivo and in vitro studies suggested that disease onset/progression is independent of the intrinsic SOD1 activity.
From the hitherto identified 119 mutations in the human SOD1 gene, more than 10 transgenic mouse lines have been established, and three of them (SOD1G93A, SOD1G85R, and SOD1G37R) have been used extensively to study ALS pathology experimentaly7.
Disturbed neuronglia signaling leads to an impaired synaptic transmission and neurotrophic support, triggering neuronal cell death in a number of circumstances for instance ALS, prion diseases, Parkinson's disease, Alzheimer's disease, etc. Studies on transgenic mouse in vivo and in vitro have been conducted to investigate whether neuron-specific expression of mutant SOD1 (mSOD1) protein is sufficient to produce an ALS phenotype or whether it might depend on the presence of mutant protein in glial cells8,9.
In fact, evidence has been obtained that accumulation of mSOD1 in postnatal motoneurons is not sufficient to induce or accelerate neuronal death in ALS mice. Recent experiments have shown that the presence of non-neuronal cells, harboring SOD1 protein, in proximity to motoneurons appears to be a prerequisite for selective neuronal death, at least in later stages of the disease. However, the underlying molecular mechanisms that lead to the impairment of neuronglia signaling in the ALS have only been partially demonstrated10.
In view of the above descriptions, methods to study glial influence on motoneuron death in ALS should be optimized. We presented in this paper our experience in obtaining non-neuronal cells to study ALS at cellular and molecular levels in vitro.
Methods
Swiss mice and also transgenic mice expressing the human SOD1 (hSOD1) gene with a G93A mutation11 were purchase from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained as hemizygotes by mating transgenic males with B6SJLF1/J females. Non-transgenic littermates were used as controls (wild-type). The colony was derived at Jackson Laboratories from G93A mutant mice with 25 ± 1.5 copies of the human SOD1 transgene12. The mice identification (mutant or wild-type) in our colony was performed by a genotyping method.
The animals were maintained in specific pathogen-free environment, in the biotery of the University of São Paulo. The mice were kept under controlled temperature and humidity conditions with a standardized light-dark cycle (lights on at 7.00 a.m. and off at 7.00 p.m.) and free access to food pellets and tap water. The study was conducted under protocols approved by the Animal Care and Use Ethic Committee at University of São Paulo and in accordance with the Guide for the Care and Use of Laboratory Animals.
Genotyping
The DNA was extracted from mice tails by using the following protocol: 500µl of K proteinase buffer 1mg/ml (Sigma Chemicals, EUA) (TrisHCl 20mM pH 8.0, NaCl 10mM, EDTA 30mM pH8.0 e SDS 0.5%) were added to each sample of tail. The suspension was incubated at 55 to 60°C during 48 hours and then centrifuged (16,000g/10 minutes, at 4°C). The aqueous phase was replaced into a new tube and 500µl cold isopropanol were added. After gently mixing, DNA became visible and could be precipitated for centrifugation (16,000g/2 minutes, at room temperature). The DNA pellet was washed twice with 500µl ethanol 70%. After removal of ethanol and 5 minutes drying, the DNA was resolved in 50µl TE buffer (10 mM Tris-HCl pH 7.4; 1mM EDTA in deionized water). DNA samples were stored at 4°C until PCR assay. PCR assay was performed as described earlier by Rosen et al.2, by using the following primers: IMR113 (5'-ATCAGCCCTAATCCATCTGA-3') and IMR114 (5'-CGCGACTAACAATCAAAGTGA-3') to amplify SOD1h fragment IMR042 (5'-CTAGGCCACAGAA TTGAAAGATCT-3') and IMR043 (5'-GTAGGTGGAAATTCTAGCATCATCC-3') to amplify a fragment of murin interleucin-2 as positive control. For each sample, PCR assay included 12.5µl PCR Master Mix (2X) (Fermentas Life Sciences), 0.5µl of each primer described above, 1µg DNA and water free from nucleotides for a final volume of 25µl, submitted to an initial period of 3 minutes at 95ºC, followed by 36 PCR amplification cycles (30 seconds at 95ºC, 30 seconds at 60ºC and 45 seconds at 72ºC) and finally 2 minutes at 72°C. PCR products with 1µl blue green could be visualized in an 1% agarosis gel.
Behavior analysis by means of hanging wire test
Neuromuscular strength was tested by the hanging wire test13. Each mouse was placed on the wire lid of a conventional housing cage. The lid was gently turned upside down, 30 cm above a soft surface to avoid injuries. The latency to fall was timed. Each mouse was given up to three attempts to hold on the inverted lid for a maximum of 180 seconds and the longest period was recorded.
Primary cultures of spinal cord cells
Astrocyte culture
The protocol used for primary spinal cord astrocyte cultures is a combination of previously protocols published8 and modification according to our experience. Post-natal day 0 (within 24 hours of birth) mouse pups were decapitated. The skin and muscles overlying the spinal cord were dissected. Spinal cord between mid-cervical and lower lumbar levels was cut and quickly cleaned of meninges in ice-cold Hank's modified balanced salt solution (HBSS; LCG Biotechnology), and the spinal cord tissue from 10 animals was pooled in fresh ice cold HBSS. The tissue was triturated with a Pasteur pipette and subsequently passed through 19G needle in Dulbecco's modified eagle medium (DMEM; Cultilab) containing 10% fetal bovine serum (FBS; Gibco), and 100U/ml penicillin and 100U/ml streptomycin (Sigma). The cell suspension was pelleted at 900 r.p.m. for 5 minutes, the supernatant was removed and the pellet resuspended in fresh DMEM at 1ml per dissected cord segment. The resuspended cells were plated onto 6-well culture plates, and maintained at 37°C in a 5% CO2/95% air atmosphere. The cultures were fed every second day with fresh DMEM. After 2 weeks, the cells were agitated (200 r.p.m. for 6 hours) to eliminate residual microglia and oligodendrocytes, then astrocytes were detached by 0.25% trypsin (Gibco) and plated onto coverslips.
Microglial culture
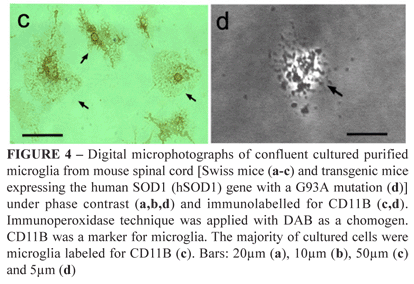
Mouse mixed glial cultures were prepared as previously described14 and adapted by our group. Spinal cords from 1 or 2-day-old neonatal mice were dissected, carefully stripped of their meninges and digested with 0.25% trypsin (Gibco) for 30 minutes at 37ºC. Trypsinization was stopped by adding an equal volume of culture medium (DMEM F12 nutrient mixture, FBS 10%, penicillin 100U/ml and streptomycin 100µg/ml) to which 0.02% deoxyribonuclease I (Sigma) was added. The solution was pelleted, re-suspended in culture medium and brought to a single cell suspension by repeated pipetting followed by passage through a 19G needle. Cells were seeded at a density of 250,000 cells/ml (¼ 62,500 cells/cm2) and cultured at 37ºC in humidified 5% CO2-95% air. Medium was replaced every 4-5 days. Cultures reached confluency after 7-10 days in vitro. After, mixed glial cultures have been confluent for at least 3 days. Mixed glial cells have been washed for 1 minute in DMEM F12 to eliminate serum. After this, the cells were incubated at 37ºC with 500µl per well of trypsin 0.25%: DMEM F12 1:3 until intact layer is detached. This step takes 4 hours. Next, 500µl of DMEM F12 with 10% fetal bovine serum was added for trypsin inactivation and the medium containing the layer of detached cells was aspirated and replaced for biochemical analysis. Microglial adherent cells could be grown15, and incubated with trypsin 0.25% and replated onto coverslips for imunohistochemical and biochemical analysis.
Immunocytochemistry
One-color immunohistochemistry
Cell cultures were rinsed 2 times with phosphate buffered saline (PBS), and then fixed in paraformaldeyde (30 minutes, at 4ºC). Sequential incubation of the fixed cells then took place at room temperature in: (1) wash in PBS for 5 minutes in agitation; (2) wash in PBS with 1% Triton for 10 minutes; (3) wash in PBS with 0.02% Tween 20 (BioRad) for 5 minutes; (4) wash in PBS with 0.02% Tween 20 and 1% BSA (Sigma) for 5 minutes; (5) block in PBS with 2% normal goat serum (NGS), 4% BSA, 0.2% Triton (Sigma) for 30 minutes; (6) primary antibody incubation: using rabbit polyclonal glial fibrillary acidic protein (GFAP), a protein of the intermediate filament of the astroglial cytoskeleton (Dako) at 1:150 dilution for astrocytes cultures, CD11B (marker for microglia, BD Biosciences) at 1:600 dilution for microglial cultures in 2% NGS, 4% BSA, 0.2% Triton in PBS overnight. (7) wash 3 times with PBS; (8) secondary antibody incubation with biotinylated anti-rabbit IgG (Vector Laboratories) at 1:250 dilution in PBS with 0.2% Triton, 1% NGS, 2% BSA for 2 hours except for microglial cultures that were incubated with a primary biotinylated antibody; (9) wash 3 times with cold PBS; (10) Incubation with avidin-biotin at 1:125 dilution in PBS; (11) wash again 3 times with PBS; (12) DAB reaction until color observation. The reaction was made in 0.01% H2O2, 0.5mg DAB/ml. DAB labeled cultures were washed 3 times with distillated water, and the reaction was interrupted with Trizma.
Two-color immunohistochemistry
Furthermore, two-color immunoperoxidase was used for further evaluation of the simultaneous detection of the GFAP (marker for astrocytes) and CD11B (marker for microglia) immunoreactivities in the astroglial cultures.
Astroglial cultures were submitted to GFAP immunocytochemistry as described above employing the DAB as a chomogen, which gave a brownish color to the immunoreactive product. Following the DAB reaction, the plated cells were rinsed several times in PBS and incubated over 24 hours at 4°C with the CD11B antiserum described above diluted 1:600. After incubated with the avidin-biotin solution (both diluted 1:250, Vectastain, Vector) for 45 minutes at room temperature, and stained for 10 minutes using 0.05% 4-chloro-naphthol (Sigma) as a chromogen and 0.05% (v/v) H2O2 (Sigma). The plated cells were rinsed in PBS and cover-slipped in a glycerol phosphate buffered medium. The sections were analyzed and photographed with an AX70 Olympus photomicroscope (U.S.A).
Western blot analysis of cultured cells
GFAP, IBA1 and chondroitin sulfate proteoglycan (NG2) protein levels were assessed by Western blot analysis. The astroglial and microglial cell cultures were homogenized in lysis buffer containing 1% protease inhibitor cocktail (Sigma), 1% NP40 (Sigma), 0.5% sodium deoxycholate (Sigma), 1% sodium dodecyl sulfate (Sigma), 1mM ethylenediaminetetraacetic acid (Sigma), 1mM ethylene glycol tetraacetic acid (Sigma), diluted in phosphate buffered saline (PBS, pH 7.4) and then homogenized. The homogenate was centrifuged (14,000r.p.m.) for 20 minutes at 4ºC; supernatants were transferred into new tubes and stored at -70ºC until use. After centrifugation at 14,000 r.p.m. for 20 minutes, the supernatants were transferred into new tubes and stored at -70ºC until use. Protein concentrations were determined according to the method described by Bradford16.
To label a GFAP and NG2 bands, the samples were separated on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (20µg of protein/lane) using a 12% TrisHCl ready gel (Bio-Rad, California, USA) at 100V for 1 hour. For to NG2, the samples were separated on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (80µg of protein/lane) using a 6% TrisHCl ready gel (Bio-Rad, California, USA) at 100V for 1 hour. Proteins were transferred to nitrocellulose membrane for 1 hour at 100V. After 1hour blocking with 5% milk in Tris-buffered saline 0.05% Tween 20 (TBS-T). Membrane was incubated overnight with rabbit polyclonal antibodies to GFAP (1:10,000, DAKO) and to NG2 (1:500, Chemicon), the antisera were diluted in 3% milk/TBS-T. Membrane was washed 2 times for 10 minutes in TBS-T and incubated at room temperature for 1 hour with anti-rabbit a 1:10,000 dilution of anti-rabbit IgGECL conjugated secondary antibody (Santa Cruz Biotechnology, USA).
For to IBA1 band, the samples were separated on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (80µg of protein/lane) using a 15% TrisHCl ready gel (Bio-Rad, California, USA) at 130V for 40 minutes. Proteins were transferred to PVDF membrane for 40 minutes at 50V. After 24 hours blocking with 5% BSA in Tris-buffered saline 0.05% Tween 20 (PBS-T). Membrane was incubated for 1 hour with rabbit polyclonal antibodies to IBA1 (1:2,000, WAKO), the antisera were diluted in PBS-T. Membrane was washed 2 times for 10 minutes in PBS-T and incubated at room temperature for 1 hour with anti-rabbit a 1:10,000 dilution of anti-rabbit IgGECL conjugated secondary antibody (Santa Cruz Biotechnology, USA).The IBA1 protein is a 17kDa protein consisting of 147 amino acids. The genomic copy of the IBA1 gene was mapped to within the major histocompatibility complex MHC class III region between the BAT2 and TNF- genes. Using a specific antibody, IBA1 protein was shown to be expressed in monocytic cell lines and microglia among the cultured brain cells. IBA1 protein functions as an adapter molecule that mediates calcium signals in the monocytic lineage, including microglia.
Blots were washed two times with TBS-T and once TBS. After final washes, the membrane was incubated with Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Science, USA) for 1 minute. The membranes were exposed to an X-ray film for imaging (HyperfilmTM ECL, Amersham Biosciences, USA) to visualize protein bands.
Gene expression by means of RT-PCR
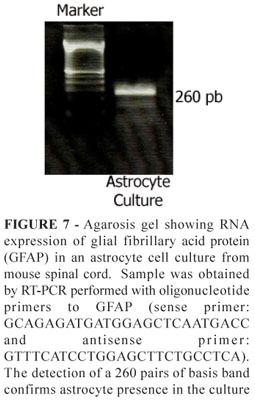
RT-PCR was performed with oligonucleotide primes 5'-GCAGAGATGATGGAGCTCAATGACC-3' and 3'-GTTTCATCCTGGAGCTTCTGCCTCA-5'. Total RNA was extracted by Trizol method. Firstly, 1ml Trizol solution was added to the cell sample, and then 200µl chloroform were added. After mixing, the phases were separated by centrifugation (12,000g/15 minutes at 4°C). The aqueous phase was transferred to a new tube and 500µl isopropanol was added. After mixing and incubation for 10 minutes at room temperature, RNA was precipitated by centrifugation (13,000g/10 minutes at 4°C). The precipitate was washed with 75% ethanol and centrifuged (1,500g/5 minutes at 4°C). After removal of the ethanol and 15 minutes drying, the RNA was resolved in 50µl RNAse free water. Complementary DNA (cDNA) templates were prepared from 1µl of total RNA with 2µl RNAse free water and Kit Ready-to-go RT-PCR beads (GE healthcare), according to the manufacturer's recommendations, followed by 40 PCR amplification cycles (94°C for 30 seconds, annealing at 60°C for 60 seconds and extension at 72°C for 90 seconds). Ten µl of PCR product was analyzed by 2% agarose gel with 2µl of ethidium bromide.
Data analysis
The Student t-test was performed to compare data from behavior analysis of the different groups of animals. Data were presented as mean ± S.E.M. and significance level was set at p<0.05.
Results
Genotyping
The animals were identified as mutant or wild-type by genotyping. These assignments were successfully performed in all mice DNA samples, and were validated by agarose gel electrophoresis of PCR products. The genotype result was determined by analysis of the bands observed. Detection of two bands was clearly observed in mutant mice B6SJL-TgN(SOD1-G93A)1 Gur with 236 pb and 324 pb. Wild-type mice showed one band with 324 pb (Figure 1).
General aspects and behavior
Animals were examined daily in their cages by a trained observer. Around the age of the 105 days, the first clinical signs of an abnormal and slow gait appeared in mutant animals. Additionally, mice showed slight trembling of the hind limb when they were suspended by their tails during normal cage maintenance. These signs were observed in both females and males mice whereas wild-type littermate controls never displayed those motor symptoms. The majority of transgenic mice was such around 125 days of age and reached the endpoint 20 days later. At death, mutant animals showed a complete paralysis.
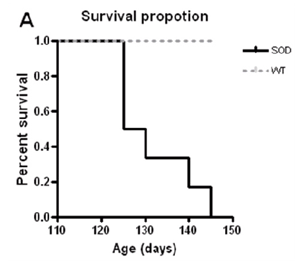
Disease onset is when these signs appear and will extend to symmetrical paralysis of the hindlimbs, followed by a complete paralysis at the end stage. Mice were killed when they were unable to roll over within 20 seconds after being pushed on their side, and this time point was recorded as the time of death. Survival of our transgenic mice is illustrated in Figure 2A.
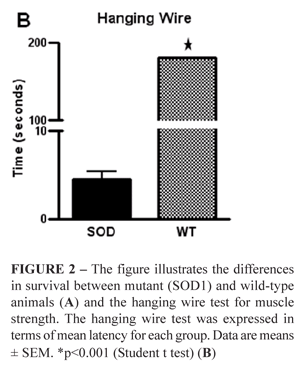
The performance on hanging wire test showed more evidenced differences between transgenic and wild-type animals (around 120 days). During the entire period of analysis all control animals continued to perform at the maximum level (Figure 2B).
Immunocytochemistry
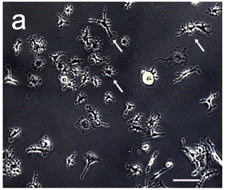
A high number of putative cultured astroglial cells was confluent and occupied the majority of the spaces in the plate surface as seen under phase contrast microscopy (Figure 3a). Immunocytochemistry demonstrated the presence of a strong labeling of GFAP in the cytoplasm and processes of the cultured astroglial cells (Figure 3b). All astrocytes in the culture stained for GFAP immunoreactivity. Confluent astrocytes possessed a round to elliptic shape with 2 long well visible processes (Figure 3b). Moreover, the cultures of astroglial cells stained for CD11B immunoreactivity demonstrated the presence of only very few and scattered microglia in the astroglial cultures (less that 1%) (Figure 3c).
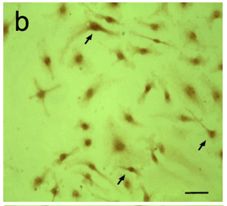
The analysis of microglial cultures of the spinal cord mice demonstrated a high number of putative cultured microglial cells that were confluent and occupied the majority of the spaces in the plate surface as seen under phase contrast microscopy (Figure 4a). Already under phase contrast, it was possible to see a number of highly ramified microglial processes of second and third order surrounding each cell (Figure 4b,d). Immunocytochemistry demonstrated the presence of a strong labeling of CD11B in the membrane of the cell cytoplasm and process of the cultured microglial cells (Figure 4c). All microglial cells in the culture stained for CD11B. Confluent microglia with their processes possessed a round to elliptic shape (Figure 4c,d). Moreover, the cultured microglial cells stained for GFAP immunoreactivity showed no immunoreaction (data not shown).
Western blot analysis
Western blot technique revealed that GFAP protein, an astroglial protein, level is more intense in the purified astroglial culture than purified microglial culture (Figure 5). No GFAP band was detected in the supernatant cells derived from the purification process of microglial cell cultures. Furthermore, the IBA1 protein, a microglial protein, level is more intense in the purified microglial culture than purified astrocyte culture. However, the western blot revealed the band of the NG2 protein, a glycoprotein present in oligodendrocyte/astrocyte precursors, only in astrocytes cultures, that have been either or not purified (Figure 5).
Two-color immunoperoxidase for simultaneous visualization of astrocytes and microglia in culture plates
Two-color immunoperoxidase method for simultaneous detection of GFAP and CD11B immunoreactivities in primary cultured astrocytes from mouse spinal cord demonstrated the presence of a large number of brownish color (DAB as a chromogen) immunoreactive astrocytes and scattered bluish color immunoreactive microglia (4-chloro-naphthol as achromogen) in the plate (Figure 6).
Control cultures incubated with the solvent for the primary antibodies, secondary antibody, or for avidin-biotin showed no reaction (data not shown).
Gene expression
The RNA of astrocyte culture cells was isolated and then transformed in cDNA by reverse transcriptase and amplified using GFAP primers. The cDNA was detected by agarose gel electrophoresis of PCR products. The reverse-transcriptase-PCR result showed a GFAP specific band of 260 pb (Figure 7).
Discussion
We have introduced the colony of transgenic mice model of ALS and the control animals for the disease in the biotery of the University of São Paulo Medical School. The animals were obtained from a specialized company abroad in producing genetically modified strains for experimental studies. We have followed the recommendations of the sender regarding amplification of the colonies of transgenic mice expressing the human SOD1 (hSOD1) gene with a G93A mutation11. The animals were maintained as hemizygotes by mating transgenic males with B6SJLF1/J females. The transgenic littermates possessed a number of copy of the human SOD1 transgene that is adequate to trigger disease onset by post natal day 105 as observed in our colonies in agreement to descriptions of groups that have used the same type of animals bearing the mutation4. Genotyping was used to detect or exclude the mutations in the littermates. Non-transgenic littermates that were called wild-type mice were used as controls. We have also used Swiss mice to compare the results regarding colony amplification, mortality and behavioral impairments as well as results obtained in the bench.
The animals were maintained in a specific pathogen-free environment in our biotery, being submitted to periodical bacteriological and proctoparasitological survey which may favor the colony amplification and homogeneity of results.
The mortality rate and longevity of G93A transgenic mice were in agreement to that described in the literature with a complete disease accomplishment onset around post natal day 125 and endpoint by 145 days of age. The average of 20-40 days of disease evolution and termination already confirms the fast progression of neurological impairments which culminate to respiratory failure and death11.
There are several tests in the literature that have been used to follow voluntary motor deterioration in animal model of ALS i.e. rotarod test, hanging wire test and electromyography. In the present paper we have employed the hanging wire test that seems to be very sensitive to demonstrate disease onset. From the behavioral results demonstrated herein, we can confirm that genotyping can predict disease onset at early adult life in hundred percent of animals in accordance to the literature13.
We have also revised in this paper the method to obtain primary culture of astrocytes8 and microglia15 which have been adapted and optimized by our group for neuroglial cells from spinal cord of mice for in vitro cellular, biochemical and molecular analyses.
Based on the methodological descriptions presented in this paper, we obtained primary cultures of mouse spinal cord astrocytes with more than ninety-five percent purity which is considered a good index for in vitro studies8,15. The phase contrast microscopy of the live cultured astrocytes showed cells with adequate vitality and morphological shape, resembling plated astrocytes. Cytological phenotype was confirmed by immunocytochemical labeling for GFAP, the intermediate filament of astroglial cytoskeleton, after paraformaldehyde fixation of astroglial primary cultures. Furthermore, the CD11B immunolabeling of fixed astroglial cultures revealed the presence of very few and scattered immunopositive microglia in the plate, thus confirming that the method leads acquisition of a remarkably purified astroglial culture.
As observed for astroglial cultures, we acquired primary cultures of mouse spinal cord microglia with more than ninety-five percent purity which is considered an excellent index for in vitro studies with that type of cell8. Phase contrast microscopy showed healthy live cultured microglia possessing morphological shape of so described plated cells15. The qualitative analysis based on cytological phenotype was confirmed by immunocytochemical labeling for CD11B, a cell surface receptor complement described with the method in the fixed cells. Furthermore, the GFAP immunolabeling of fixed microglial cultures revealed the presence of very few and scattered immunopositive astrocytes in the plate, thus prompting the acquisition and expansion of a purified microglial culture.
We have also demonstrated by means of Western blot technique that the obtained primary neuroglial cultures allow biochemical evaluation of protein contents in the plated cells after ressupension, homogenization and protein extraction which is very useful in the quantitative studies of molecular signaling that participates in the pathophysiological mechanisms of the ALS disease17. We have employed the Western blot analysis to further characterize the identity and purity of astroglial and microglial cultures. Purified microglial cultures showed a strong 17 kDA band of IBA1 immunoblot and almost no detectable 50 kDA GFAP band. In the other way, purified astroglial cultures showed an intense GFAP band and a slight IBA1 band, indicating a well acceptable degree of purity of both cell types by means of a biochemical method. The method was also employed to detect NG2, a marker of oligodendrocyte/protoplasmic astrocyte precursors, in both non-purified and purified astrocytes, but not in microglial culture, an indication that those precursor cells are displaying mitogenic activity in culture. Moreover, it is of substantial interest the observation of the absence of GFAP, IBA1 and NG2 in the seeded supernatant cells after confluence from the process of microglial culture purification. That finding does emphasize the necessity of a good method to acquire cell culture purification18, because other type of cells, probably endothelial, ependimal and meningeal fibroblast cells, are present in the initial phases of the process and showed be eliminated. As far as we know, this is the first time the degree of purity has been detailed in the described methods to obtain those primary cultured non-neuronal cells derived from mouse spinal cord.
We have also demonstrated in this paper the RNA expression of GFAP in purified mouse astrocyte cultures by means of RT-PCR technique. Besides the importance of the method to characterize the plated cells, RT-PCR can be employed for molecular analysis of contribution of non-neuronal cells in the pathophysiological mechanisms of ALS progression.
Conclusion
It is possible to achieve highly purified primary cultures of spinal cord astrocytes and microglia to be employed in cellular and molecular analyses of the influence of such non-neuronal cells in the pathophysiology of ALS.
Acknowledgement
The authors thank to Instituto Paulo Gontijo for the incentive.
Received: December 14, 2009
Review: February 12, 2010
Accepted: March 18, 2010
Conflict of interest: none
Financial source: FAPESP (98/13122-5; 99/01319-1; 07/00491-3) and CNPq
- 1. Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol. 1998;24(2):104-17.
- 2. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59-62.
- 3. Shaw PJ. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry. 2005;76(8):1046-57.
- 4. Julien JP, Kriz J. Transgenic mouse models of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762(11-12):1013-24.
- 5. Andersen PM. The genetics of amyotrophic lateral sclerosis (ALS). Suppl Clin Neurophysiol. 2004;57:211-27.
- 6. Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39-59.
- 7. Schymick JC, Talbot K, Traynor BJ. Genetics of sporadic amyotrophic lateral sclerosis. Hum Mol Genet. 2007;16 Spec No. 2:R233-42.
- 8. Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007;10(5):615-22.
- 9. Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillée S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302(5646):113-7.
- 10. Jaarsma D, Teuling E, Haasdijk ED, De Zeeuw CI, Hoogenraad CC. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci. 2008;28(9):2075-88.
- 11. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772-5.
- 12. Gurney ME. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci. 1997;152 Suppl 1:S67-73.
- 13. Miana-Mena FJ, Munoz MJ, Yague G, Mendez M, Moreno M, Ciriza J, Zaragoza P, Osta R. Optimal methods to characterize the G93A mouse model of ALS. Amyotroph Lateral Scler 2005;6:55-62.
- 14. Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6(8):2163-78.
- 15. Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44(3):183-9.
- 16. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-54.
- 17. Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500(1):20-46.
- 18. Duobles T, Lima Tde S, Levy Bde F, Chadi G. S100beta and fibroblast growth factor-2 are present in cultured Schwann cells and may exert paracrine actions on the peripheral nerve injury. Acta Cir Bras. 2008;23(6):555-60.
Publication Dates
-
Publication in this collection
21 May 2010 -
Date of issue
June 2010
History
-
Reviewed
12 Feb 2010 -
Received
14 Dec 2009 -
Accepted
18 Mar 2010