Resumos
OBJETIVO: Relatar dois pacientes, um acometido por trissomia do cromossomo 13 em mosaico e outro por trissomia do cromossomo 18, ambos com apresentações clínicas não usuais. DESCRIÇÃO DO CASO: Paciente do sexo feminino de dois meses de idade, que apresentava deficiência de crescimento, dismorfias menores de face e de membros, paresia facial unilateral, cardiopatia congênita, hipotonia e evolução com o surgimento de manchas hipocrômicas e atraso do desenvolvimento neuropsicomotor. O segundo caso é de um paciente do sexo masculino, com 19 dias de vida, que também mostrava deficiência de crescimento, anomalias faciais menores, defeito radial, cardiopatia congênita e hipertonia. Os cariótipos por bandas GTG confirmaram o diagnóstico, respectivamente, de síndromes de Patau e de Edwards. COMENTÁRIOS: Os presentes relatos têm por objetivo alertar os pediatras sobre manifestações não usuais nas trissomias dos cromossomos 13 e 18, as quais podem dificultar a suspeita diagnóstica.
trissomia; cromossomos humanos par 13; mosaicismo; cromossomos humanos par 18
OBJECTIVE: Report two patients, one with trisomy 13 mosaicism and the other with trisomy 18, both with unusual clinical presentations. CASE DESCRIPTION: The first case was a female patient with two months of age who presented growth deficiency, minor dysmorphia of face and limbs, unilateral facial paresis, congenital heart defect and hypotonia, who developed hypochromic spots and neuropsychomotor delay. The second case was a male patient with 19 days of age who also had growth deficiency, minor facial anomalies, radial defect, congenital heart defect and hypertonia. GTG-Banding karyotypes confirmed the diagnosis of Patau and Edwards syndromes respectively. COMMENTS: The aim of these reports is to call attention to the fact that Patau and Edwards syndromes may present an unusual pattern of abnormalities, making the diagnostic hypothesis difficult.
trisomy; chromosomes human pair 13; mosaicism; chromosomes human pair 18
RELATO DE CASO
Apresentações clínicas não usuais de pacientes portadores de síndrome de Patau e Edwards: um desafio diagnóstico?
Unusual clinical presentations of patients with Patau and Edwards syndromes: a diagnostic challenge?
Paulo Ricardo G. ZenI; Rafael Fabiano M. RosaII; Rosana Cardoso M. RosaIII; Lisiane Dale MulleIV; Carla GraziadioV; Giorgio Adriano PaskulinVI
IGeneticista clínico da Universidade Federal de Ciências da Saúde de Porto Alegre (UFCSPA) e do Complexo Hospitalar Santa Casa de Porto Alegre (CHSCPA), mestre pelo Programa de Pós-graduação em Genética e Biologia Molecular da Universidade Federal do Rio Grande do Sul (UFRGS) e professor-assistente da disciplina de Genética Clínica da UFCSPA. Porto Alegre, RS, Brasil
IIGeneticista clínico da UFCSPA e do CHSCPA e mestre pelo Programa de Pós-graduação em Patologia da UFCSPA. Porto Alegre, RS, Brasil
IIIMédica formada pela UFCSPA. Porto Alegre, RS, Brasil
IVMédica intensivista pediátrica do Hospital da Criança Santo Antônio do CHSCPA. Porto Alegre, RS, Brasil
VGeneticista clínica da UFCSPA e do CHSCPA, mestre pelo Programa de Pós-graduação em Genética e Biologia Molecular da UFRGS e professora assistente da disciplina de Genética Clínica da UFCSPA. Porto Alegre, RS, Brasil
VIGeneticista clínico da UFCSPA e do CHSCPA, doutor pelo Programa de Pós-graduação em Genética e Biologia Molecular da UFRGS e professor adjunto da disciplina de Genética Clínica da UFCSPA. Porto Alegre, RS, Brasil
Endereço para correspondência Endereço para correspondência: Paulo Ricardo Gazzola Zen Rua Sarmento Leite, 245, sala 403 - Centro CEP 90050-170 Porto Alegre/RS E-mail: paulozen@ufcspa.edu.br
RESUMO
OBJETIVO: Relatar dois pacientes, um acometido por trissomia do cromossomo 13 em mosaico e outro por trissomia do cromossomo 18, ambos com apresentações clínicas não usuais.
DESCRIÇÃO DO CASO: Paciente do sexo feminino de dois meses de idade, que apresentava deficiência de crescimento, dismorfias menores de face e de membros, paresia facial unilateral, cardiopatia congênita, hipotonia e evolução com o surgimento de manchas hipocrômicas e atraso do desenvolvimento neuropsicomotor. O segundo caso é de um paciente do sexo masculino, com 19 dias de vida, que também mostrava deficiência de crescimento, anomalias faciais menores, defeito radial, cardiopatia congênita e hipertonia. Os cariótipos por bandas GTG confirmaram o diagnóstico, respectivamente, de síndromes de Patau e de Edwards.
COMENTÁRIOS: Os presentes relatos têm por objetivo alertar os pediatras sobre manifestações não usuais nas trissomias dos cromossomos 13 e 18, as quais podem dificultar a suspeita diagnóstica.
Palavras-chave: trissomia; cromossomos humanos par 13; mosaicismo; cromossomos humanos par 18.
ABSTRACT
OBJECTIVE: Report two patients, one with trisomy 13 mosaicism and the other with trisomy 18, both with unusual clinical presentations.
CASE DESCRIPTION: The first case was a female patient with two months of age who presented growth deficiency, minor dysmorphia of face and limbs, unilateral facial paresis, congenital heart defect and hypotonia, who developed hypochromic spots and neuropsychomotor delay. The second case was a male patient with 19 days of age who also had growth deficiency, minor facial anomalies, radial defect, congenital heart defect and hypertonia. GTG-Banding karyotypes confirmed the diagnosis of Patau and Edwards syndromes respectively.
COMMENTS: The aim of these reports is to call attention to the fact that Patau and Edwards syndromes may present an unusual pattern of abnormalities, making the diagnostic hypothesis difficult.
Key-words: trisomy; chromosomes human pair 13, mosaicism; chromosomes human pair 18.
Introdução
As síndromes de Patau e de Edwards, ambas descritas em 1960(1,2), são doenças genéticas bastante conhecidas e causadas, respectivamente, pela trissomia do cromossomo 13 e 18. Essas doenças apresentam prevalência na população de 1:10.000-20.000 e 1:3.600-8.500 nascidos vivos, respectivamente, com predomínio, nas duas síndromes, de meninas. A síndrome de Edwards representa a segunda trissomia mais freqüente de cromossomos autossômicos, ultrapassada apenas pela síndrome de Down(3-6) .
Ambas caracterizam-se por um quadro clínico amplo, com acometimento de múltiplos órgãos e sistemas. A maioria dos fetos portadores de trissomia do cromossomo 13 e 18 não chega ao termo(5-7). Dos nascidos vivos, a quase totalidade (respectivamente, 86 e 90-100%) evolui para o óbito no primeiro ano de vida(3,8). Em ambas, o diagnóstico é usualmente confirmado pelo estudo dos cromossomos, com o exame de cariótipo a partir do sangue periférico.
Relatam-se aqui dois pacientes, um com síndrome de Patau e outro com síndrome de Edwards, ambos com apresentações clínicas não usuais.
Caso 1
Paciente do sexo feminino, branca, dois meses de idade, segundo filho de um casal de pais jovens e não consangüíneos. A criança foi ao hospital para avaliação genética por cardiopatia congênita, dificuldade de ganho de peso e hipotonia. Ao nascimento, ela pesava 2.730g (entre percentil P três e dez da curva de crescimento intra-uterino), estatura de 45cm (menor que P3), perímetro cefálico de 36cm (entre P50 e 98) e Apgar de dez no quinto minuto.
No exame físico, aos dois meses de idade, a paciente pesava 3.445g (menor que P3 da curva de crescimento pós-natal), media 50cm (menor que P3), perímetro cefálico de 38,5cm (P50), apresentava hipotonia, estreitamento bitemporal, fendas palpebrais oblíquas para cima, ponte nasal alargada, narinas antevertidas e micrognatia. A orelha direita era pequena, mas de tamanho não inferior ao P3, com ausência da sua curvatura superior e presença de uma fosseta pré-auricular à altura do trago. A comissura labial mostrava-se desviada para a esquerda, secundária a uma paresia facial (Figura 1A e 1C). Nas mãos, evidenciava-se clinodactilia e hipoplasia da falange média do quinto dedo. Hemangiomas planos, um na glabela e outro acima do mamilo direito, bem como sopro cardíaco sistólico e uma pequena fosseta na região lombo-sacra também estavam presentes (Tabela 1).
Na ecocardiografia, observou-se comunicação interatrial (CIA) e interventricular (CIV). A avaliação ultra-sonográfica do abdome foi normal e a ressonância nuclear magnética cerebral revelou ventrículos laterais de dimensões aumentadas.
O estudo citogenético pelo cariótipo por bandas GTG em leucócitos mostrou a presença de mosaicismo envolvendo duas linhagens celulares: uma portadora de uma translocação equilibrada entre os cromossomos 13 e 14 e de uma inversão pericêntrica na região heterocromática de um dos cromossomos do par 1; outra com as mesmas alterações e um cromossomo 13 adicional {mos 45,XX,inv(1)(p11q21),der(13;14)(q10;q10)[35]/46,XX,inv(1)(p11q21),der(13;14)(q10;q10),+13[5]}. O estudo citogenético dos pais não foi realizado por opção dos mesmos.
A paciente evoluiu com atraso de desenvolvimento neuropsicomotor (DNPM), apesar de conseguir atingir os marcos do desenvolvimento: andou sem apoio aos 28 meses e pronunciou os primeiros dissílabos com 25 meses (Figura 1C). Aos três anos de idade, ainda apresentava atraso pôndero-estatural, com peso e altura abaixo de P3. Além disso, os pais perceberam, a partir de um ano de idade, o surgimento de manchas hipocrômicas em forma de listras na região do tronco e dos membros, que seguiam as linhas de Blaschko (Figura 1B).
Caso 2
Trata-se de um paciente do sexo masculino, branco, com 17 dias de vida, único filho de pais jovens e não consangüíneos, com história de dois abortos espontâneos prévios. A criança nasceu de parto cesáreo, apresentação cefálica, com 36 semanas de gestação, pesando 2.400g (entre P10 e P50 da curva de crescimento intra-uterino), medindo 44cm (P10) e com Apgar de dez no quinto minuto. A avaliação ultra-sonográfica fetal evidenciou polidrâmnio, peso reduzido, cardiopatia e alteração de membros. O recém-nascido evoluiu com disfunção respiratória e septicemia, necessitando de ventilação mecânica. A avaliação ecocardiográfica detectou a presença de comunicação interventricular e interatrial persistência do canal arterial e válvula aórtica e pulmonar bicúspides.
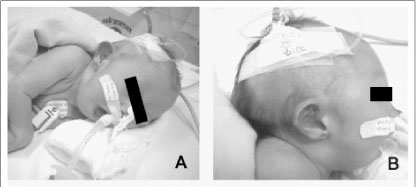
No exame físico, com 19 dias de vida, apresentava peso de 2.100g (menor que P3 da curva de crescimento pós-natal), comprimento de 46cm (menor que P3), perímetro cefálico de 33cm (P2), hipertonia, fendas palpebrais pequenas e oblíquas para baixo, raiz nasal alta, boca pequena, micrognatia, orelha direita proeminente com anti-hélice apagada e occipital proeminente (Figura 2A e 2B). Na ausculta cardíaca, evidenciou-se sopro sistólico. Nas extremidades superiores, verificou-se ausência de polegar à esquerda, sendo o mesmo pedunculado à direita, com desvio radial bilateral de mãos e antebraços. Constatou-se ainda ausência de pregas nos dedos das mãos e presença de fossetas na face ulnar e distal dos antebraços (Figura 2C). Nas extremidades inferiores, os pés apareciam em mata-borrão, com calcâneos proeminentes e fossetas na face medial das pernas, em seus terços médios, junto às tíbias (Figura 2D).
O estudo radiológico evidenciou agenesia de rádio à esquerda e hipoplasia à direita, ulnas hipoplásicas e encurvadas, polegar ausente à esquerda e séssil à direita, com luxação congênita bilateral do quadril. No exame de ultra-som abdominal observou-se rins de tamanho simétrico, com aumento da ecogenicidade cortical e, no ultra-som cerebral, ventrículos laterais proeminentes (Tabela 2). O paciente teve piora do quadro clínico geral, indo a óbito com 31 dias de vida.
O resultado do exame de cariótipo com bandamento GTG demonstrou a presença de um cromossomo 18 adicional (47,XY,+18), confirmando o diagnóstico de síndrome de Edwards por trissomia livre do cromossomo 18.
Discussão
O nascimento de uma criança com a síndrome de Patau ou de Edwards geralmente se associa a uma situação de muita ansiedade, tanto por parte dos pais do paciente como dos diferentes profissionais da área da saúde.
A síndrome de Patau, em geral, resulta da presença de cromossomo 13 adicional (trissomia livre), levando a uma constituição citogenética com 47 cromossomos(4). Esta se origina do fenômeno de não-disjunção, na maioria das vezes durante a segunda divisão meiótica da oogênese materna e se associa à idade da mãe. Aproximadamente 20% dos casos são decorrentes de translocações, na sua maioria Robertsonianas, resultantes da fusão de dois cromossomos acrocêntricos, em especial entre os cromossomos 13 e 14, como ocorreu no presente caso. Esta representa o principal tipo de translocação Robertsoniana (cerca de 33% dos casos), com uma prevalência estimada de portadores na população de 1:1.300 indivíduos(9). Nestes casos, o estudo dos cromossomos dos pais é sempre indicado no intuito de excluir a possibilidade de que um deles possa ser portador equilibrado desta alteração e apresentar, assim, um risco aumentado para sua prole. Somente 5% dos casos de síndrome de Patau ocorrem por mosaicismo, ou seja, pela presença de mais de uma linhagem de células no mesmo organismo, como no caso da paciente (3).
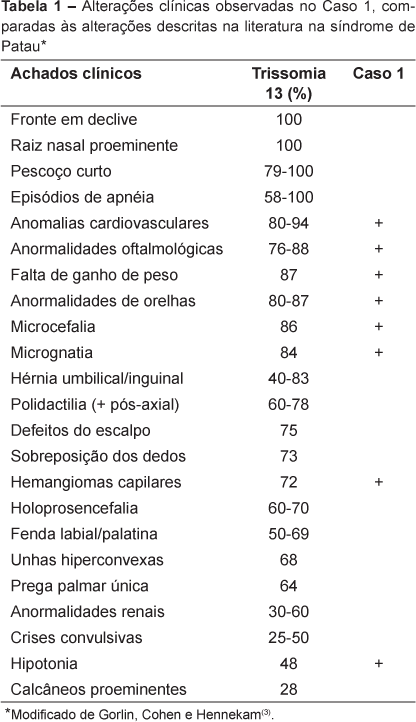
A trissomia do cromossomo 13, ao contrário da trissomia do cromossomo 18, apresenta mais freqüentemente malformações evidentes ao exame físico, que constituem padrão característico de múltiplas anomalias congênitas. Em geral, a combinação de fendas orofaciais (como lábio leporino), microftalmia e/ou anoftalmia e polidactilia pós-axial de membros permite seu reconhecimento(4). Essa tríade característica é observada em 60 a 70% dos casos (Tabela 1).
Nos casos de mosaicismo, o fenótipo tende a ser menos comprometido(3), contudo, há pequena correlação entre o fenótipo e o grau de mosaicismo verificado nos linfócitos(4). Esta observação foi condizente com o quadro clínico da paciente (Tabela 1), sendo que o achado de manchas hipocrômicas seguindo as linhas de Blaschko é sugestivo da presença deste mosaicismo. Tal associação é raramente descrita na literatura e, em alguns casos, pode haver necessidade da análise cariotípica de outros tecidos (como a pele) para a confirmação diagnóstica(10-12).
A expectativa média de vida dos pacientes com trissomia do cromossomo 13 é de 130 dias, sendo a sobrevida além dos três anos de idade excepcional(3). As crianças que ultrapassam esta faixa etária apresentam, de forma geral, importante atraso no desenvolvimento, mas com capacidade de adquirirem algumas habilidades, como ocorreu com a paciente(4).
A inversão envolvendo a região heterocromática do cromossomo 1, observada na avaliação citogenética do presente caso, é um polimorfismo constitucional (ou seja, uma variante normal da população), não se associando a anormalidades fenotípicas nos portadores(13).
Os pais da criança foram orientados quanto à possibilidade de que um deles pudesse ser portador não só da mesma inversão pericêntrica do cromossomo 1, como também da translocação entre os cromossomos 13 e 14 identificadas na paciente, além dos possíveis riscos que tal alteração significaria para futuras gestações. Contudo, os mesmos optaram por não realizar o exame de cariótipo, o que impossibilitou um adequado aconselhamento genético. Naqueles casos em que a translocação é identificada, o risco observado é inferior a 1% devido à grande seleção intra-útero(6).
A síndrome de Edwards, em geral, resulta de uma trissomia livre, ou seja, da presença de três cópias inteiras do cromossomo 18. Tal trissomia também possui uma associação com a idade da mãe, sendo a maioria dos casos decorrente de um fenômeno de não-disjunção durante a segunda divisão meiótica da oogênese materna. Translocações e mosaicismo são menos freqüentes, sendo observados em somente 10% dos casos(3,4).
O padrão sindrômico da trissomia do cromossomo 18 também é bastante distinto e raramente confundido com outras condições. Marion et al(14) desenvolveram um sistema de escores para otimizar a sua identificação. Contudo, dentre as características apresentadas pelo paciente, a que mais chamou atenção e dificultou o seu reconhecimento, foi a anormalidade radial de membros superiores. A presença desta anomalia é incomum na síndrome de Edwards, sendo observada em apenas 5 a 10% dos casos(4). A alteração de membros mais freqüente e muito sugestiva da síndrome é a camptodactilia dos dedos das mãos (Tabela 2).
Nos casos de trissomia livre do cromossomo 18 não existe indicação de realizar a avaliação citogenética dos pais, pois esta anormalidade, como já exposto, é decorrente de um fenômeno de não-disjunção durante a gametogênese. Alguns autores sugerem um discreto aumento do risco em futuras gestações, mesmo para outras trissomias potencialmente viáveis, sendo que certas mulheres apresentariam uma predisposição a um número maior de erros meióticos em geral. Contudo, a recorrência da mesma trissomia em outro filho tem sido raramente relatada na literatura, sendo seu risco considerado virtualmente desconhecido. Entretanto, deve-se lembrar do aumento do risco de concepção de fetos trissômicos com o avanço da idade materna(9).
Assim, os presentes relatos têm por objetivo expor e alertar que doenças genéticas clássicas, como a síndrome de Patau e de Edwards, podem apresentar um padrão não usual de anormalidades, tornando seu diagnóstico um desafio. Nesses casos, o cariótipo é essencial para a sua definição. O diagnóstico preciso destas síndromes é fundamental para avaliação, conduta clínica e aconselhamento genético adequados.
Recebido em: 8/1/2008
Aprovado em: 25/5/2008
- 1. Patau K, Smith DW, Therman E, Inhorn SL, Wagner HP. Multiple congenital anomaly caused by an extra chromosome. Lancet 1960;1:790-3.
- 2. Edwards JH, Harnden DG, Cameron AH, Crosse VM, Wolff OH. A new trisomic syndrome. Lancet 1960;1:787-90.
- 3. Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the head and neck. 4th ed. New York: Oxford University Press; 2001.
- 4. Cassidy SB, Allanson JE. Management of genetic syndromes. 2nd ed. New Jersey: Wiley-Liss; 2005.
- 5. Sugayama SMM, Kim CA, Leone CR, Diniz EMA, Koiffmann CP, Gonzalez CH. História natural de 24 pacientes com trissomia 18 (síndrome de Edwards) e de 20 pacientes com trissomia 13 (síndrome Patau). Pediatria (São Paulo) 1999;21:69-77.
- 6. Sugayma SMM, Kim CA, Albano LMJ, Utagawa CY, Bertola DR, Koiffmann CP et al Estudo genético-clínico de 20 pacientes com trissomia 13 (síndrome de Patau). Pediatria (São Paulo) 1999;21:21-9.
- 7. Simpson JL, Elias S. Genetics in obstetrics and gynecology. 3rd ed. Philadelphia: Saunders; 2003.
- 8. Jones KL. Smith's recognizable patterns of human malformation. 6th ed. Philadelphia: Elsevier; 2006.
- 9. Gardner RJM, Sutherland GR. Chromosome abnormalities and genetic counseling. 3rd ed. New York: Oxford University Press; 2004.
- 10. Pillay T, Winship WS, Ramdial PK. Pigmentary abnormalities in trisomy of chromosome 13. Clin Dysmorphol 1998;7:191-4.
- 11. Happle R. Phylloid hypomelanosis is closely related to mosaic trisomy 13. Eur J Dermatol 2000;10:511-2.
- 12. Yakinci C, Kutlu NO, Alp MN, Senol M, Durmaz Y, Budak T. Hypomelanosis of ito with trisomy 13 mosaicism [46, XY, der (13;13) (q10;q10), +13/46,xy]. Turk J Pediatr 2002;44:152-5.
- 13. Verma RS, Ramesh KH, Mathews T, Kleyman SM, Conte RA. Centromeric alphoid sequences are breakage prone resulting in pericentromeric inversion heteromorphism of qh region of chromosome 1. Ann Genet 1996;39:205-8.
- 14. Marion RW, Chitayat D, Hutcheon RG, Neidich JA, Zackai EH, Singer LP et al Trisomy 18 score: A rapid, reliable diagnostic test for trisomy 18. J Pediatr 1988;113:45-8.
Datas de Publicação
-
Publicação nesta coleção
29 Set 2008 -
Data do Fascículo
Set 2008
Histórico
-
Aceito
25 Maio 2008 -
Recebido
08 Jan 2008