Resumos
Terapia gênica é o tratamento baseado na introdução de genes sadios com uso de técnicas de DNA recombinante. O primeiro teste clínico bem-sucedido dessa técnica foi divulgado em 1990. Em que pese a ocorrência, em certos estudos clínicos, de efeitos adversos, alguns dos quais graves, laboratórios de pesquisa e empresas vêm continuamente desenvolvendo novos materiais e procedimentos mais seguros e eficazes. Embora ainda em estágio experimental, progressos recentes indicam oportunidades crescentes de investimento pela indústria, bem como justificam a expectativa de que, em alguns casos, essa tecnologia poderá chegar à prática clínica dentro de poucos anos.
Genes; Doenças genéticas; Engenharia genética; Manipulação do genoma; Terapias avançadas; Biotecnologia
Gene therapy is the therapeutic procedure based on the introduction of healthy genes using recombinant DNA techniques. The first successful clinical trial of this technique was published in 1990. Despite the occurrence, in certain clinical trials, of adverse effects, some of which serious, both laboratories and companies are continuously developing novel materials and establishing both safer and more effective procedures. Although still in experimental stages, recent progress both points to growing opportunities for investment by industry, as well as justify the expectation that, in some cases, this technology may reach clinical practice within a few years.
Genes; Genetic diseases; Genetic engineering; Genome manipulation; Advanced therapies; Biotechnology
DOSSIÊ BIOTECNOLOGIA
Terapia gênica: o que é, o que não é e o que será
Rafael Linden
RESUMO
Terapia gênica é o tratamento baseado na introdução de genes sadios com uso de técnicas de DNA recombinante. O primeiro teste clínico bem-sucedido dessa técnica foi divulgado em 1990. Em que pese a ocorrência, em certos estudos clínicos, de efeitos adversos, alguns dos quais graves, laboratórios de pesquisa e empresas vêm continuamente desenvolvendo novos materiais e procedimentos mais seguros e eficazes. Embora ainda em estágio experimental, progressos recentes indicam oportunidades crescentes de investimento pela indústria, bem como justificam a expectativa de que, em alguns casos, essa tecnologia poderá chegar à prática clínica dentro de poucos anos.
Palavras-chave: Genes, Doenças genéticas, Engenharia genética, Manipulação do genoma, Terapias avançadas, Biotecnologia.
ABSTRACT
Gene therapy is the therapeutic procedure based on the introduction of healthy genes using recombinant DNA techniques. The first successful clinical trial of this technique was published in 1990. Despite the occurrence, in certain clinical trials, of adverse effects, some of which serious, both laboratories and companies are continuously developing novel materials and establishing both safer and more effective procedures. Although still in experimental stages, recent progress both points to growing opportunities for investment by industry, as well as justify the expectation that, in some cases, this technology may reach clinical practice within a few years.
Keywords: Genes, Genetic diseases, Genetic engineering, Genome manipulation, Advanced therapies, Biotechnology.
Introdução
DESDE sua fundação, pelo monge Johann (Gregor) Mendel no século XIX, aos dias de hoje, a genética evoluiu extraordinariamente e conquistou um lugar de destaque entre as ciências. Há dez anos foi completado o sequenciamento do genoma humano (Lander et al., 2001; Venter et al., 2001), um feito grandioso que promete acelerar o progresso da biologia e da medicina do século XXI.
A medicina moderna acrescenta, a cada dia, descobertas importantes em áreas de investigação destinadas ao desenvolvimento de novos paradigmas de tratamento para doenças ainda incuráveis. Entre elas, a expectativa de curar doenças genéticas repousa sobre a identificação de genes responsáveis por sua patogênese e sobre o avanço das tecnologias de DNA recombinante, ou "engenharia genética", que permitem a manipulação do genoma de forma cada vez mais eficiente e segura (Watson et al., 2006). Em paralelo, a determinação de fatores genéticos de suscetibilidade a certas doenças, seu curso e suas manifestações clínicas (NCBI, 2009), bem como o enorme avanço na compreensão da biologia celular e molecular de eventos patológicos fundamentais, tais como processos inflamatórios, distúrbios de proliferação e morte celular programada (Coleman & Tsongalis, 2009), aumentam a expectativa de que a manipulação do genoma possa vir a ser aplicada a uma ampla gama de doenças.
Essa é uma área ainda incipiente da medicina, praticada especialmente nos laboratórios de pesquisa fundamental, e sua aplicação ainda é estritamente experimental. Já há nessa área produtos comerciais aprovados para uso médico (Pearson et al., 2004), mas a expectativa dos cientistas, bem como da indústria farmacêutica e de biotecnologia, é de que a liberação de protocolos de manipulação do genoma para a prática médica e o respectivo mercado de biológicos deverão avançar cautelosamente ao longo dos próximos 5-10 anos, ainda assim englobando um número restrito de aplicações.
Já em 1990, entretanto, uma equipe médica norte-americana tinha inserido um gene sadio no organismo de uma menina doente e a criança melhorou após esse tratamento. Começara uma nova era. A era da terapia gênica (ou terapia genética), ou seja, o procedimento destinado a introduzir em um organismo, com o uso de técnicas de DNA recombinante, genes sadios (nesse contexto denominados "genes terapêuticos") para substituir, manipular ou suplementar genes inativos ou disfuncionais (Linden, 2008).
Primórdios da terapia gênica
A partir da década de 1940, a genética tomou grande impulso, e descobertas sobre a natureza, composição química e as propriedades do material genético, bem como as primeiras manipulações do DNA de bactérias, começaram a gerar expectativas de novos avanços terapêuticos.
Em meados da década de 1960, começou a especulação sobre a possibilidade de utilizar vírus para transferir genes a seres humanos doentes e curar doenças genéticas (Friedmann, 1997). Já naquela época, considerava-se tanto que os próprios genes de certos vírus pudessem fazer efeito quanto que fosse possível inserir genes humanos sadios em vírus para que esses os transferissem ao paciente. Entretanto, foi só no início da década seguinte que Paul Berg conseguiu de fato manipular uma molécula de DNA (Jackson et al., 1972), criando a tecnologia do DNA recombinante.
Duas tentativas iniciais de aplicar na prática clínica o conceito de terapia gênica fracassaram, uma delas por se apoiar em uma premissa sobre propriedades de um vírus, a qual, mais tarde, se mostrou falsa (Rogers, 1952; Rogers & Rous, 1951; Andrewes, 1966; Friedman, 2001; Scaglia & Lee, 2006); outra, embora tecnicamente justificável e já utilizando metodologias de DNA recombinante, foi maculada por grave deslize ético (Mercola & Cline, 1980). Mas, em 1989, um novo teste, feito de acordo com as regras vigentes na época, restabeleceu expectativas positivas nessa área de pesquisa.
A paciente tratada em 1989 era uma menina de quatro anos de idade incapaz de levar uma vida normal, porque sofria de uma doença genética causada por deficiência da enzima adenosina desaminase (ADA), indispensável para o desenvolvimento do sistema imune. Várias mutações no gene que codifica a enzima provocam deficiência de ADA, o que resulta em degeneração das células T do sistema imune (Buckley, 2004) e constitui uma das principais causas de síndrome de imunodeficiência combinada severa (SCID, do inglês severe combined immunodeficiency). No caso em questão, a doença é conhecida pela sigla SCID-ADA. Crianças afetadas pelas diversas formas de SCID (ibidem) têm baixíssima resistência a infecções e, se não forem tratadas, morrem em geral antes dos seis meses de idade. São conhecidas como "crianças da bolha", por necessitarem de isolamento feito, frequentemente, por meio de compartimentos de plástico transparente. O tratamento é usualmente feito por reposição da enzima através de injeções semanais. Naquele caso, depois de um período de um ano em que houve relativo sucesso, no segundo ano de tratamento a criança voltou a sofrer infecções frequentes e desenvolveu uma alergia ao preparado da enzima usado para injeções. Os indícios eram de que a terapia de reposição enzimática estava falhando. O médico William French Anderson, da Universidade do Sul da Califórnia, obteve então autorização dos comitês de ética para iniciar um teste de terapia gênica (Anderson et al., 1990).
A cada um ou dois meses, os pesquisadores retiravam células T do sangue de Ashanti, inseriam o gene da ADA, induziam a proliferação dessas células no laboratório e, então, devolviam as células tratadas para o sangue da paciente (Culver et al., 1991). Depois de sete infusões, houve uma pausa de seis meses, e, a partir daí, as infusões recomeçaram até o tratamento completar dois anos. Por segurança, a menina continuou a receber as injeções semanais da enzima. A terapia gênica dessa paciente, bem como a realizada a partir de 1991 em uma segunda paciente de nove anos de idade, teve resultados positivos. Houve melhora clínica com uma redução da quantidade de enzima que era necessário repor. Observou-se que os níveis da enzima no sangue das pacientes aumentaram progressivamente com a terapia gênica e se mantiveram estáveis no intervalo de descanso de seis meses (Blaese et al., 1995; Mullen et al., 1996). Finalmente, doze anos após terminarem as infusões, época em que foi feita uma reavaliação dos dois casos, grandes números de células T continuaram expressando o gene terapêutico no sangue da primeira paciente, cujo tratamento foi mais bem-sucedido do que o da segunda (Muul et al., 2003).
Deve-se assinalar que ainda há questões técnicas relacionadas a esse estudo, que não permitem considerá-lo um completo sucesso clínico. Como as crianças continuaram a receber reposição da enzima, embora em doses menores, há dúvida sobre o quanto a terapia gênica terá de fato contribuído para que, por exemplo, a primeira paciente esteja hoje, aos 24 anos de idade, saudável e ativa. No entanto, a partir das observações feitas ao longo do tratamento dessas duas primeiras pacientes, a terapia gênica para SCID-ADA evoluiu e hoje é considerada um sucesso clínico (Aiuti et al., 2009; Kohn & Candotti, 2009). Mesmo incipiente, o estudo iniciado em 1989, que obteve pelo menos alguns resultados positivos observando os requisitos éticos, é um marco na história da terapia gênica e inspirou o crescimento subsequente dessa área de investigação científica.
Modalidades de terapia gênica
A ideia de usar as técnicas de DNA recombinante para corrigir o genoma foi inspirada nas doenças causadas por mutação em um único gene (ditas doenças monogênicas). Nesse caso, a ideia é substituir ou suplementar a expressão do gene disfuncional, mediante a inserção de uma ou mais cópias do gene terapêutico (Porteus et al., 2006; O'Connor & Crystal, 2006; Brinkman et al., 2006). O tratamento da SCID-ADA representa uma aplicação bem-sucedida dessa ideia.
Mas as doenças monogênicas não são o único alvo da terapia gênica (Figura 1). A medicina moderna luta contra muitas doenças complexas, cujas causas primárias ainda não são conhecidas e para as quais há, na melhor das hipóteses, apenas tratamentos paliativos. Em certos casos, é possível planejar uma intervenção por meio de terapia gênica, visando reduzir ou evitar a progressão da doença. A intervenção pode ser baseada no conhecimento de determinantes genéticos de suscetibilidade ou gravidade, ou na oportunidade de alterar mecanismos fundamentais ou a fisiologia das células, dos órgãos ou sistemas afetados pelas doenças (Cardone, 2007; Flotte, 2007). As principais estratégias são aumentar a resistência celular, estimular sistemas de reparo ou regeneração, ou ainda recompor características funcionais específicas de determinados sistemas orgânicos, mediante modulação de genes não necessariamente associados à causa da doença (Bagley et al., 2008; Lundberg et al., 2008). Já no caso de tumores, o principal objetivo é a indução de morte celular seletiva em populações celulares proliferativas (Bauzon & Hermiston, 2008; Cattaneo et al., 2008; Ribacka et al., 2008).
Finalmente, há uma forma peculiar de terapia gênica denominada vacina de DNA. Nessa, ao invés da utilização de uma proteína ou um vírus completo inativado, como se faz nas vacinas convencionais, o paciente recebe o gene que codifica uma proteína típica do agente agressor. Dessa forma, o organismo do paciente passará a fabricar permanentemente a proteína exógena, estimulando seu próprio sistema imune. Essas vacinas podem ter finalidade preventiva, de forma semelhante às vacinas clássicas, ou curativa, levando o sistema imune a atacar os agentes agressores já instalados no organismo (Atkins et al., 2008, Sykes, 2008; Silva et al., 2009).
Terapia celular, células-tronco e terapia gênica
As células-tronco são, atualmente, o principal assunto de natureza médica na mídia. Ao mesmo tempo, criou-se certa confusão entre células-tronco, terapias celulares e terapias genéticas. Nas chamadas terapias celulares, empregam-se células inteiras para tratar uma doença, com base nas propriedades regenerativas de células-tronco ou em outros efeitos, a maior parte dos quais ainda não explicados, das células transplantadas. O exemplo clássico, cuja fundamentação é bem conhecida, é o de leucemias, mas há expectativa de que muitas classes de doenças possam vir a ser tratadas com emprego de terapias celulares nos próximos anos (Torrente & Polli, 2008; Gribben, 2008; Einstein & Ben-Hur, 2008; Reffelmann et al., 2008).
No presente contexto, é importante frisar que terapias celulares não envolvem necessariamente modificação genética. Já as terapias gênicas são baseadas na introdução ou modificação de genes. Isso pode ser feito diretamente in vivo, sem o auxílio de células inteiras do próprio paciente ou de doadores.
Ou seja, terapia gênica e terapia celular são dois conceitos distintos. Entretanto, há métodos que combinam as duas técnicas. Um exemplo de combinação de terapia gênica com terapia celular foi, novamente, o procedimento ex vivo que inaugurou a terapia gênica, e que foi descrito antes. Novas tecnologias de terapia gênica para a SCID-ADA são baseadas na manipulação genética de células-tronco de medula óssea, em lugar das células T empregadas nos primeiros estudos (Aiuti et al., 2009). Portanto, em certas circunstâncias, podem-se utilizar células como veículo para introduzir o gene terapêutico. Mas são a introdução do gene e o uso das tecnologias de DNA recombinante que caracterizam o tratamento como terapia gênica.
Vetores para terapia gênica
A base da terapia gênica consiste na introdução de genes em células. Porém, a entrada de DNA puro através da membrana plasmática de células eucarióticas é extremamente rara (Vellai & Vida, 1999). Essa dificuldade é, naturalmente, benéfica para o organismo, pois dificulta alterações espúrias do metabolismo celular e até mesmo transformações semelhantes às que se observam na evolução das espécies.
Por conseguinte, de modo geral, há necessidade de um carreador que facilite a entrada do DNA nas células vivas. Esse veículo é denominado "vetor". Há três classes principais de vetores atualmente em desenvolvimento: plasmídeos, vetores virais e vetores nanoestruturados.
Plasmídeos
Os plasmídeos são sequências de DNA relativamente simples, porém eficazes para expressão de genes, nas quais é possível inserir um gene terapêutico por técnicas de DNA recombinante (Voss, 2007; Clanchy & Williams, 2008; Gill et al., 2009). Mas, para vencer a resistência das células à introdução de plasmí-deos, é preciso fragilizar a membrana celular, o que pode ser obtido por diversos métodos, como o emprego de choques elétricos ou substâncias que fragilizam quimicamente a membrana celular (Dass, 2004; Cemazar & Sersa, 2007; Favard et al., 2007; Wu & Lu, 2007). Outra alternativa consiste em aplicar uma grande quantidade de plasmídeos nas vizinhanças das células, de modo que, mesmo com eficiência muito baixa, uma pequena fração que seja capaz de cruzar a membrana já produza efeitos, ou ainda injetar rapidamente um grande volume de solução contendo plasmídeos (Herweijer & Wolff, 2007).
Essas técnicas são, entretanto, muito limitadas. Por exemplo, é improvável seu uso para introduzir genes em órgãos de difícil acesso, como o cérebro. Assim, o emprego de vetores plasmidiais é limitado a algumas circunstâncias, tais como sua introdução por injeção intramuscular, como no caso das vacinas de DNA ou no músculo cardíaco, ou ainda em estudos experimentais em animais. Outrossim, essa tecnologia pode ter aplicações importantes, por exemplo, para introduzir o gene sadio em células isoladas e combinar terapia gênica com terapia celular.
Vetores virais
Em contraposição à resistência da membrana celular à entrada espontânea de DNA em uma célula, os vírus são micro-organismos especializados exatamente em invadir células e nelas introduzir material genético. Contêm ácido nucleico (DNA ou RNA) cercado por uma capa de proteína e, em alguns casos, de um envelope adicional de proteína e lipídeos e seu ciclo de vida implica liberação do ácido nucleico viral na célula hospedeira. Essa propriedade é explorada para introduzir genes terapêuticos nas células, por meio de tecnologias de DNA recombinante.
Alguns vetores são derivados de adenovírus. Essa família inclui quase 50 tipos distintos de vírus que causam, por exemplo, faringites ou conjuntivites. Infecções por adenovírus são muito comuns, e, por isso, a maior parte da população possui anticorpos contra um ou mais tipos dessa família de vírus. Outros são da família dos retrovírus, que inclui o HTLV causador de um tipo de leucemia e o HIV causador da Aids, que pertence à subfamília dos lentivírus, os quais vêm sendo muito estudados como fonte de vetores para terapia gênica. Ainda outros vetores são derivados de vírus da família dos adenovírus-associados, que não são patogênicos para seres humanos.
O princípio da produção de vetores de origem viral para terapia gênica (figuras 2 e 3) consiste em remover os genes envolvidos nos mecanismos patogênicos e de proliferação viral, mantendo apenas o necessário para invasão das células sem multiplicação, seguida da inserção de um gene terapêutico no que resta do DNA viral (Machida, 2002). A remoção de genes que conferem o caráter patogênico e a multiplicação permite, por exemplo, que um vírus da mesma subfamília do perigoso HIV possa dar origem a um vetor viral útil para terapia gênica.
Os vetores virais diferem entre si (Tabela 1). Uns são mais eficientes, outros têm maior capacidade de veicular genes grandes. Alguns têm maior propensão a provocar reações inflamatórias do que outros. Finalmente, alguns vetores, como os derivados de retrovírus, têm a propriedade de se integrar ao genoma das células. Isso é positivo quando se quer uma expressão permanente do gene terapêutico, mas pode causar efeitos adversos graves.
Vetores nanoestruturados
Outra forma de introduzir DNA em células está sendo desenvolvida a partir de preparados obtidos por técnicas avançadas de nanotecnologia (Sanvicens & Marco, 2008). Aí se incluem polímeros que formam verdadeiras redes que prendem um gene e soltam sua carga quando penetram nas células, bem como vesículas de lipídeos contendo o DNA, capazes de fundir com a membrana das células, liberando seu conteúdo no interior destas últimas.
Esses vetores podem ser enriquecidos com moléculas que ajudem a especificar em que tipos de células o conteúdo poderá penetrar, ou ainda permitam guiar ou transferir seletivamente os vetores de um compartimento para outro, por exemplo, do sangue para o cérebro (Pardridge, 2005, 2007, Figura 4). Esta última técnica é importante, pois facilitará a terapia gênica cerebral sem a necessidade de uma neurocirurgia para introduzir o vetor, bastando injeções endovenosas.
Ainda em outros casos, células modificadas pela introdução de um gene terapêutico podem ser encapsuladas em compartimentos produzidos a partir de polímeros inertes e, depois, introduzidas no organismo. A vantagem dessa técnica é que as células podem produzir e secretar moléculas terapêuticas enquanto ficam isoladas do sistema imune do paciente (Hauser et al., 2004; Lindvall & Wahlberg, 2008). Portanto, as células encapsuladas não precisam ser derivadas do próprio paciente.
Terapia gênica hoje
As terapias gênicas são procedimentos novos que ainda se encontram em fase experimental. O conhecimento básico vem sendo adquirido em laboratórios de pesquisa fundamental por meio de testes em modelos experimentais e ensaios pré-clínicos. Esses estudos validam o potencial de eficácia de uma estratégia terapêutica, bem como permitem detectar potenciais riscos a seres humanos, antecipando modificações dos vetores e outros componentes da estratégia terapêutica que aumentem a segurança para uso humano.
A pesquisa fundamental em terapia gênica é intensa e crescente no mundo. A Figura 5 ilustra o crescimento contínuo do volume de publicações científicas nessa área. Nos últimos três anos, foram publicados, em média, cerca de 30 artigos científicos sobre assuntos relacionados a terapia gênica por dia.
Como em outras áreas de investigação de novos métodos terapêuticos, a aprovação de um produto ou processo de terapia gênica depende da realização de uma série de ensaios clínicos, que são classificados por fases. Inicia-se pela chamada fase I, cujo objetivo é testar a segurança do procedimento e identificar quaisquer efeitos adversos atribuídos ao novo produto ou método. Seguem-se ensaios de fase II, III e IV que, progressivamente e sempre acompanhados de vigilância quanto a efeitos adversos, destinam-se a testar a eficácia do novo produto ou método em amostras crescentes de pacientes, frequentemente distribuídos em múltiplos centros de pesquisa.
A realização desses ensaios clínicos de terapia gênica depende de aprovação prévia por comitês de ética locais e nacionais, como a Comissão Nacional de Ética em Pesquisa (Conep) no Brasil ou a Food and Drug Administration (FDA) nos Estados Unidos. No caso de terapia gênica, existe ainda no Brasil a Comissão Técnica Nacional de Biossegurança (CTNBio) e nos Estados Unidos um comitê específico do Instituto Nacional de Saúde (NIH, do inglês National Institutes of Health), chamado RAC (do inglês Recombinant DNA Advisory Committee), que devem autorizar procedimentos envolvendo DNA recombinante. No entanto, diferentemente dos Estados Unidos, ainda não existe no Brasil regulamentação específica sobre terapia gênica, a qual precisa, urgentemente, ser elaborada tanto para evitar o uso inadequado das terapias quanto para controlar a produção e importação de insumos do exterior. No momento, resta às autoridades sanitárias aplicar normas consagradas no exterior para examinar eventuais pedidos de licença ou fiscalizar ensaios clínicos e eventuais produtos de terapia gênica no país.
Em todo o mundo, até junho de 2010 haviam sido compilados cerca de 1.650 ensaios clínicos em terapia gênica na base de dados da revista Journal of Gene Medicine (http://www.wiley.co.uk/genmed/clinical/). As figuras 6-11 ilustram os principais aspectos do estado atual da pesquisa clínica nessa área.
A distribuição dos países-sede de ensaios clínicos (Figura 6) corresponde, de modo geral, ao investimento feito na pesquisa fundamental em anos precedentes. Dentre os países componentes do grupo "outros", a base de dados do JGM inclui um ensaio sediado no México e nenhum na América do Sul. De fato, dos 38 ensaios clínicos em andamento em países da América do Sul identificados ao final de 2009 na base de dados do Instituto Nacional de Saúde dos EUA (www.clinicaltrials.gov), 37 constituem extensões de ensaios sediados em países do Hemisfério Norte e apenas um, iniciado em 2009, é de fato sediado na América do Sul, especificamente no Brasil (ver adiante).
A distribuição em fases (Figura 7) reflete claramente o caráter experimental da terapia gênica. Para comparação, podem-se citar dados do conjunto dos ensaios clínicos registrados na página clinicaltrials.gov. Dentre esses ensaios, que incluem predominantemente fármacos e procedimentos clínicos e cirúrgicos convencionais, cerca de 45% são de fase II e pouco mais de 30% são de fase III. Já, como demonstrado no gráfico da Figura 7, a maioria dos ensaios clínicos em terapia gênica ainda não passa da fase I e, até o momento, apenas cerca de 4% progrediram até as fases III e IV. Ainda assim, há sinais de que a progressão da terapia gênica experimental no sentido da prática médica está se acelerando (Figura 8).
A segurança ainda é a principal barreira ao desenvolvimento da terapia gênica para a prática médica. O principal entrave é o fato de que os vetores não virais mais seguros disponíveis no momento são ainda pouco eficientes ou têm aplicação muito limitada, como é o caso dos plasmídeos discutidos antes. A alta eficiência de transdução de vetores virais torna estes últimos os mais promissores para aplicação. Entretanto, alguns tipos, particularmente de vetores adenovirais e retrovirais, os mais utilizados até hoje, produziram efeitos adversos, alguns graves e mesmo fatais, e contribuíram fortemente para o bloqueio de muitos estudos na fase I.
Naturalmente, 150 anos de pesquisa fundamental em farmacologia oferecem uma base sólida, sobre a qual questões de segurança de medicamentos convencionais são frequentemente resolvidas nos laboratórios de pesquisa básica ou em ensaios pré-clínicos consagrados e altamente preditivos. Ainda há um longo caminho a percorrer até que essa situação se torne rotina na pesquisa em terapia gênica.
A distribuição dos ensaios clínicos por indicação terapêutica (Figura 9) corrobora um aspecto já mencionado. Embora a terapia gênica tenha sido concebida originalmente com o objetivo de tratar doenças monogênicas, essas constituem hoje o alvo de menos de 10% dos ensaios clínicos. A predominância de câncer pode ser explicada, em parte, pela maior facilidade de aprovação de ensaios clínicos baseados no uso compassionado de drogas ou terapias experimentais em pacientes terminais, mas também pelo grande avanço no desenho de vírus oncolíticos (que destroem células tumorais) e terapias com genes suicidas (ver adiante).
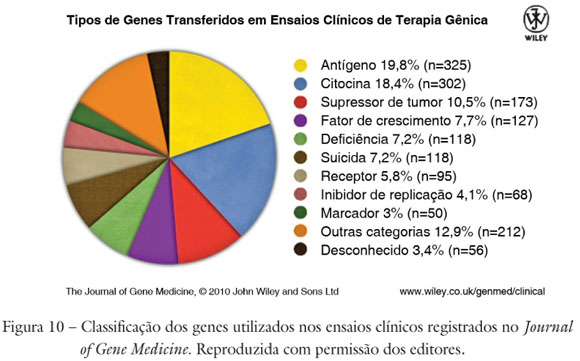
A variedade de genes utilizados nos ensaios clínicos (Figura 10) reflete o caráter ad hoc da terapia gênica. É provável que o avanço dessa área de pesquisa médica seja fortemente influenciado pela tendência ao desenvolvimento de medicina personalizada, com base em avanços da genética, da farmacogenômica e de outros campos de investigação moderna. Ainda assim, a prevalência de antígenos, citocinas, supressores de tumor e genes suicidas corresponde à predominância de câncer como indicação terapêutica mais frequente na pesquisa clínica nessa área.
O tópico de vetores é, sem dúvida, o mais crítico para o avanço da terapia gênica no sentido da aplicação à prática médica. O gráfico da Figura 11 compila dados obtidos em duas décadas, ao longo das quais o progresso tecnológico na área de vetores foi extraordinário. Por exemplo, em contraposição ao tipo de vetor adenoviral que ensejou a morte de um paciente de um ensaio clínico em 1999 (ver adiante) e quase paralisou a pesquisa em terapia gênica, hoje estão disponíveis vetores adenovirais de terceira geração, radicalmente modificados no sentido de evitar efeitos adversos como o que vitimou aquele paciente. Cresce, contudo, a expectativa de utilização de vetores virais intrinsecamente mais seguros, como os vetores derivados de vírus adenoassociado.
Aplicações da terapia gênica
Para ilustrar as aplicações potenciais da terapia gênica, bem como a lógica subjacente e a sequência da pesquisa fundamental e pré-clínica que levou aos ensaios clínicos, foram selecionados alguns exemplos ilustrados a seguir.
Doenças monogênicas
Hemofilia: Como cada tipo de hemofilia é uma doença monogênica, o procedimento é o de introduzir o respectivo gene sadio (fator VIII ou fator IX, dependendo do tipo de hemofilia) em células do paciente, para que essas passem a produzir a proteína necessária. A terapia deve não apenas fazer o organismo voltar a produzir a proteína que falta, mas produzi-la em quantidade suficiente para restabelecer a saúde do paciente e por longo prazo, idealmente por toda a vida.
Após extensos ensaios pré-clínicos em camundongos e cães, que demonstraram recuperação de longo prazo da atividade pró-coagulante mediada por fator IX introduzido por terapia gênica experimental, dois estudos de fase I/II foram realizados recentemente por um grupo da Universidade da Pensilvânia, com aplicação de um vetor derivado de vírus adenoassociado (rAAV), contendo o gene codificante do fator IX em pacientes de hemofilia B (Manno et al., 2003, 2006; Hasbrouck & High, 2008). Não houve efeitos adversos sérios em nenhum dos pacientes testados.
Os resultados indicam potencial eficácia do tratamento, pois um paciente que recebeu uma dose elevada do rAAV-F9 por infusão hepática apresentou, entre duas e cinco semanas após o tratamento, níveis terapêuticos de fator IX circulante acima de 10% da atividade normal, que é suficiente para sustentar a capacidade de coagulação sanguínea. Entretanto, o efeito terapêutico foi transitório, desaparecendo seis semanas após o tratamento, acompanhado de aumento temporário e assintomático de níveis de transaminases (Figura 12). Os resultados deste e de outro paciente no mesmo estudo indicaram que os efeitos terapêuticos foram abolidos por degeneração das células do fígado nas quais foi introduzido o vetor, causada por uma resposta imune contra proteínas do vetor viral (Mingozzi & High, 2007).
Esse exemplo é particularmente importante, porque em nenhum dos ensaios pré-clínicos realizados em animais antes da formulação do estudo clínico, e nem mesmo em novos experimentos realizados após a obtenção dos resultados do ensaio clínico citado, foram observadas nos animais de experimentação respostas imunitárias que permitissem prever a resposta imune observada nos pacientes. O resultado demonstra a necessidade de cautela na transição de estudos pré-clínicos para ensaios clínicos, mesmo na ausência de efeitos adversos sérios, e forneceu dados cruciais para o avanço da aplicação clínica de terapia gênica. Novos estudos experimentais estão em andamento, visando evitar essa resposta imunitária com o emprego de variantes do vetor e imunossupressão transitória, que guiarão novos ensaios clínicos (Hasbrouck & High, 2008).
Amaurose congênita de Leber: A partir do final de abril de 2008, foram também divulgados os primeiros resultados de ensaios clínicos de fase I/II para tratamento da amaurose congênita de Leber (abreviada LCA, do inglês Leber's congenital amaurosis). A LCA é uma doença que provoca cegueira progressiva, iniciando-se com perda importante de visão em bebês e progredindo ao longo do tempo para cegueira total. Inicialmente, os fotorreceptores, células retinianas sensíveis à luz e imprescindíveis para a visão, são inativados, mas permanecem vivos na retina (den Hollander et al., 2008). Com o passar dos anos, os fotorreceptores inativos, predominantemente os bastonetes que funcionam em baixos níveis de luminosidade, degeneram e desaparecem (Spuy et al., 2005).
Há várias formas de LCA, algumas de causa genética já bem conhecida, como a deficiência da RPE65, uma enzima necessária para produzir o derivado de vitamina A essencial para o funcionamento dos fotorreceptores (Poehner et al., 2000; Bereta et al., 2008). Os fotorreceptores desses pacientes perdem paulatinamente a função, mas sua degeneração só costuma acontecer por volta dos 30 anos de idade (Hollander et al., 2008). Esse curso da doença oferece uma janela terapêutica para inserção de cópias normais do gene que codifica a RPE65 na retina de adultos jovens portadores desse tipo de LCA (Figura 13). Os testes são ainda preliminares, e, em princípio, somente três pacientes foram testados em cada um de três ensaios clínicos de fase I realizados na Inglaterra e nos Estados Unidos (Bainbridge et al., 2008; Maguire et al., 2008; Cideciyan et al., 2008; Hauswirth et al., 2008).
Os primeiros resultados mostraram que a introdução dos vetores derivados de adenovírus-associado contendo o gene normal na retina dos pacientes não provocou efeitos adversos importantes. Foi observada melhora em exames oftalmológicos e no desempenho visual de alguns doentes, que recuperaram parcialmente a sensibilidade à luz (Hauswirth et al., 2008) e a capacidade de se orientar em ambientes de baixa luminosidade, o que não conseguiam fazer antes da introdução do gene sadio (Bainbridge et al., 2008).
Os resultados até agora, entretanto, foram obtidos em poucos pacientes, ainda não foram observados sinais de melhora em certos testes oftalmológicos cruciais (Hauswirth et al., 2008) e a deficiência de RPE65 é responsável por apenas 6% dos casos de LCA (Hollander et al., 2008). Ou seja, a terapia que está em teste no momento, se for bem-sucedida, só poderá ser aplicada a uma fração pequena dos doentes. Tratamentos para os demais grupos de pacientes terão de ser desenvolvidos caso a caso. Ainda assim, trata-se de um avanço importante no desenvolvimento de novas terapias para doenças que levam à cegueira, e já está em andamento um ensaio clínico fase II para confirmar (ou não), de forma sistemática, a possível eficácia do tratamento.
Câncer
A maioria dos ensaios clínicos de terapia gênica tem sido feita em pacientes de câncer (Figura 9), em geral em estágios avançados. O efeito desejável de qualquer tratamento para o câncer é o de provocar a morte seletiva das células tumorais (Evan & Littlewood, 1998; Green & Evan, 2002). Células cancerosas geralmente multiplicam-se com rapidez, o que explica o crescimento dos tumores. Muitos fármacos são usados no tratamento do câncer justamente porque atacam seletivamente células que se multiplicam com rapidez e, portanto, matam células tumorais (Wang et al., 2008; Prochownik, 2008; Vazquez et al., 2008).
A necessidade fisiológica de renovação contínua das células do sangue, a partir da proliferação de precursores na medula óssea, implica, entretanto, efeitos adversos graves da quimioterapia. Esses efeitos são difíceis de evitar, pois, entre outros fatores, os medicamentos são injetados na circulação. Para tratamento de câncer, é desejável atingir, de alguma forma, apenas as células tumorais. No caso de tumores sólidos, como tumores originados no sistema nervoso central, isso é possível mediante terapia gênica localizada (Rainov & Ren, 2003), e várias estratégias vêm sendo desenvolvidas nesse sentido (Tabela 2).
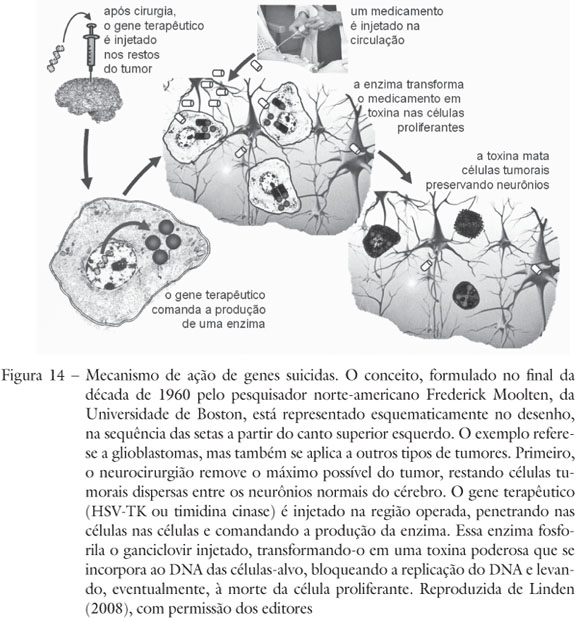
O procedimento apelidado de "técnica de genes suicidas" consiste em introduzir nas células tumorais um gene que não existe no genoma humano e codifica a enzima timidina cinase, proveniente do genoma do herpesvírus. A presença dessa enzima em uma célula humana mata a célula na presença de uma droga chamada ganciclovir, pois a timidina cinase transforma o ganciclovir em uma toxina. A toxina, por sua vez, só afeta células que se multiplicam (Figura 14).
Embora a eficácia da tecnologia de gene suicida para tratamento de tumores seja ainda controversa, alguns estudos obtiveram resultados animadores. Dentre eles, um ensaio clínico de fase I/II realizado na Finlândia, no qual a ressecção de tumores extremamente agressivos do sistema nervoso central, denominados glioblastomas, foi seguida por injeção, no leito cirúrgico, de um vetor adenoviral contendo o gene da timidina cinase de herpesvírus. O procedimento prosseguiu com injeções endovenosas diárias de ganciclovir por 14 dias. A terapia gênica resultou em aumento significativo da sobrevida (Figura 15) do grupo de 17 pacientes tratados por terapia gênica, quando comparado a um grupo de 19 pacientes tratados com terapia convencional, ou quando comparado a uma população controle de 36 pacientes previamente tratados por métodos convencionais na mesma unidade de neurocirurgia, nos dois anos anteriores ao ensaio (Immonen et al., 2004). O vetor utilizado nesse estudo está sendo desenvolvido pela empresa Ark Therapeutics que, recentemente, relatou resultados positivos significativos de um estudo multicêntrico de fase III com 250 pacientes e, em fevereiro de 2009, obteve na França a primeira autorização para uso compassionado do produto, denominado Cerepro®.
Doença de Parkinson
As doenças neurodegenerativas são uma das classes mais problemáticas para a medicina contemporânea. Apesar dos avanços ocorridos desde os anos 1990, período denominado "década do cérebro" (Goldstein, 1994), e do extenso conjunto de conhecimentos acumulados sobre diversos aspectos da patogênese, genética, curso clínico, complicações e resposta aos diversos tratamentos testados ao longo de anos de investigação, há uma conspícua carência de opções terapêuticas, particularmente nos estágios mais avançados destas doenças (Radunovic et al., 2007; Cacabelos, 2007; Han & McDonald, 2008; Jalbert et al., 2008; Gauthier & Poirier, 2008; Olanow et al., 2008).
Por sua vez, algumas neurodegenerações são ilustrativas do potencial de desenvolvimento de terapia gênica para doenças multifatoriais e de alta complexidade. A doença de Parkinson (DP) é um exemplo dessa categoria.
A DP é caracterizada por perda progressiva de neurônios na parte compacta da substância negra do mesencéfalo e alterações funcionais em outros núcleos do tronco cerebral (Figura 16), acompanhada da formação de inclusões intracelulares denominadas corpos de Lewy. Isso resulta em depleção de dopamina, o neurotransmissor utilizado pelos neurônios que degeneram, no alvo dos prolongamentos dos neurônios da substância negra, que se chama corpo estriado. Com a evolução da doença, encontra-se adicionalmente o envolvimento de outros sistemas de neurotransmissores. Os distúrbios motores típicos da doença, como tremor de repouso, lentidão dos movimentos e rigidez muscular, são frequentemente acompanhados por instabilidade postural, disfunção visceral e distúrbios cognitivos (Guttman et al., 2003). Os mecanismos que levam à morte dos neurônios da substância negra são ainda controversos (Dawson & Dawson, 2003; Dauer & Przedborski, 2003).
O tratamento farmacológico com L-Dopa, um medicamento precursor da síntese de dopamina, é eficaz em curto ou médio prazo, mas tende a se tornar inócuo com a perda progressiva dos neurônios, além de eventualmente provocar distúrbios motores adicionais. A progressão da doença exige doses mais elevadas e combinações de medicamentos, que nem sempre se mostram eficazes (Poewe, 2009). Terapias celulares destinadas a repor neurônios dopaminérgicos na substância negra poderão, eventualmente, beneficiar pacientes de DP, mas, até o momento, os ensaios clínicos efetuados com transplantes de tecido nervoso fetal tiveram efeitos discretos, bem como sugeriram a possibilidade de transmissão da doença para o tecido transplantado (Thajeb et al., 1997; Li et al., 2008; Kordower et al., 2008; Mendez et al., 2008; Braak & Del Tredici, 2008).
Estratégias de terapia gênica para tratamento da doença de Parkinson incluem a indução da produção local de dopamina no estriado, a oferta de fatores neurotróficos para reduzir a perda progressiva de neurônios dopaminérgicos ou, ainda, a compensação do desequilíbrio funcional na rede de comunicação celular dos núcleos da base (Chen et al., 2005).
A produção de dopamina depende essencialmente da atividade de três enzimas. As técnicas destinadas a produzir dopamina no corpo estriado depletado envolvem, em geral, a indução de uma ou mais destas enzimas por meio de vetores virais (Kang et al., 2001). Os modelos experimentais pré-clínicos consistem em lesões químicas da substância negra em ratos ou em primatas. Foram testados vários tipos de vetores virais (Chen et al., 2005 para revisão). Com base nos resultados dos estudos pré-clínicos, foi iniciado um ensaio clínico fase I destinado a testar a segurança e, secundariamente, efeitos benéficos de terapia gênica por expressão de uma das enzimas produtoras (a AADC), veiculada por vetor adenoviral-associado injetado no corpo estriado de pacientes que sofrem da DP, em média, há 14 anos (http://clinicaltrials.gov/show/NCT00229736). Os resultados (Christine et al., 2009) demonstraram melhora do quadro clínico sem efeitos adversos da terapia gênica per se, embora tenham sido detectados riscos no procedimento operatório.
Por sua vez, estratégias de neuroproteção, destinadas a reduzir ou impedir a perda neuronal a longo prazo, têm sido formuladas com base em diversos fatores de crescimento que têm efeito protetor sobre neurônios da substância negra. Dentre os experimentos feitos em animais, um estudo testou os efeitos da injeção, no corpo estriado, de um vetor viral expressando uma construção do gene de neurturina, com resultados positivos (Fjord-Larsen et al., 2005). Assim, um ensaio clínico fase I foi iniciado em meados de 2005, visando examinar a segurança de um vetor viral adenoassociado expressando o gene de neurturina injetado no corpo estriado (http://clinicaltrials.gov/show/NCT00252850). Não houve efeitos adversos graves em 12 pacientes tratados com duas doses distintas do vetor, e foram detectados efeitos benéficos em alguns parâmetros motores (Marks et al., 2008). Um estudo multicêntrico de fase II encontra-se, agora, em andamento.
A terceira estratégia de terapia gênica para DP é baseada no desequilíbrio funcional entre vias excitadoras e inibidoras nos núcleos da base, consequente à perda da atividade da substância negra (Figura 16). Nessas condiçõe, ocorre desinibição da atividade de um núcleo chamado núcleo subtalâmico (STN), à qual se atribui importante papel nos principais sinais da DP (Nakano, 2000; Chen et al., 2005). Vários estudos demonstraram que remoção cirúrgica do STN ou estimulação elétrica de alta frequência tem efeitos benéficos sobre alguns desses sinais, justificando o emprego da chamada estimulação cerebral profunda no tratamento de casos avançados de DP (Diamond & Jankovic, 2005). O conhecimento das propriedades funcionais de circuitos neurais envolvidos na doença levou a um exemplo notável de intervenção genética destinada a modular a fisiologia do sistema nervoso, independentemente da causa da doença que, ainda hoje, continua controversa.
Foi desenvolvido um ensaio de terapia gênica que consiste na indução de expressão de enzimas que produzem um neurotransmissor inibidor, visando contrapor-se ao excesso de atividade neural no STN. A expressão dessas enzimas no STN produziu efeitos funcionais benéficos em modelo de DP em ratos (Luo et al., 2002). Com base nesses resultados, foi conduzido, no período 2003-2005, um ensaio clínico de fase I de terapia gênica empregando um vetor de vírus adenoassociado recombinante, contendo o gene que codifica uma dessas enzimas, injetado no STN (http://www.clinicaltrials.gov/ct/show/NCT00195143). Resultados de 11 pacientes acompanhados por até 12 meses indicaram melhora significativa de desempenho motor, acompanhada de redução de atividade metabólica em alvos de projeção do STN, compatível com os resultados dos estudos pré-clínicos. Foi também divulgada melhora significativa em escala de atividades cotidianas, que reflete a opinião dos pacientes sobre seu desempenho em tarefas do dia a dia. Não foram relatados efeitos adversos que deponham contra a segurança do procedimento (Kaplitt et al., 2007).
Os resultados dos ensaios clínicos descritos são, ainda, muito preliminares, foram obtidos em números reduzidos de pacientes e necessitam de confirmação em ensaios mais amplos, com controles mais rigorosos para efeito placebo e outras variáveis. Portanto, é ainda cedo para concluir sobre a viabilidade e, particularmente, a eficácia de terapia gênica para doenças neurodegenerativas. Entretanto, esses estudos somam-se a outros ensaios clínicos que sugerem que a terapia gênica poderá se transformar em alternativa efetiva de tratamento para doenças hoje incuráveis.
O balanço risco-benefício da terapia gênica
Dentre as centenas de ensaios clínicos de terapia gênica já encerrados, a maioria destinou-se a testar a segurança do procedimento. Em certos casos, a identificação precoce de efeitos adversos durante o estudo foi suficiente para encerrar imediatamente o teste, evitando risco de agravamento. Mas, em muitos casos, o procedimento empregado foi considerado seguro, quando muito com efeitos adversos ocasionais, discretos e toleráveis.
Dor ou inflamação leves no local da injeção, febre baixa transitória, dor de cabeça passageira, sintomas semelhantes à gripe e outros efeitos suaves são, em geral, toleráveis em vista do potencial de tratamento de uma doença incurável. Esses são a maior parte dos incidentes que se costuma encontrar nos ensaios clínicos de fase I em terapia gênica, especialmente após a realização de extensos testes pré-clínicos em animais, exigidos pelas agências reguladoras para autorização de ensaios clínicos (acesso a agências reguladoras nos Estados Unidos e Europa e regulamentação nessa área pode ser obtido, por exemplo, pelo link http://www.genetherapynet.com/legislation.html).
Reações imunitárias, entretanto, não apenas podem provocar efeitos adversos, mas, mesmo que não o façam, podem destruir os vetores ou as células infectadas por vetores virais, em que pese o uso de técnicas sofisticadas de DNA recombinante em sua fabricação. Esse foi o caso do ensaio para hemofilia do tipo B, já descrito (Mingozzi & High, 2007), mas que não trouxe consequências significativas aos pacientes. Em outros casos, no entanto, os efeitos adversos podem ser muito severos ou, em raros casos, fatais.
Em 1999, um paciente morreu logo após a injeção de um vetor viral durante um ensaio clínico de terapia gênica, vitimado por uma síndrome de resposta inflamatória sistêmica causada pelo vetor adenoviral de primeira geração (Raper et al., 1998, 2003). Em ensaios clínicos mais recentes, realizados na França e Inglaterra (Hacein-Bey-Abina et al., 2002; Gaspar et al., 2004), de um total de 20 crianças abaixo de um ano de idade submetidas a terapia gênica para síndrome de imunodeficiência combinada severa ligada ao cromossomo X (SCID-XL) (Buckley, 2004), cinco desenvolveram leucemias (Hacein-Bey-Abina et al., 2003; Howe et al., 2008). Dessas, uma foi a óbito e quatro apresentaram remissão completa da leucemia após quimioterapia. Exames feitos após o aparecimento das leucemias revelaram que os vetores retrovirais utilizados em ambos os ensaios produziram mutagênese insercional, ou seja, mutações produzidas pela intromissão do vetor no DNA, rompendo a continuidade da sequência genética (Cavazzana-Calvo & Fischer, 2007; Howe et al., 2008).
Os casos citados constituem os mais graves exemplos efetivamente caracterizados como efeitos adversos diretos da terapia gênica. Ambos têm origem em características dos vetores virais utilizados. Porém, em ambos os casos, a pesquisa fundamental, aliada à observação criteriosa dos eventos associados ao tratamento e ao curso clínico dos efeitos colaterais, contribuiu para avanços no desenho e produção de novos vetores, destinados a evitar tais efeitos adversos.
No caso de vetores adenovirais, em contraposição à primeira geração de vetores empregada no ensaio clínico que resultou no caso fatal de 1999, já estão disponíveis vetores adenovirais de terceira geração, construídos com deleção completa de genes virais e capazes de transdução gênica muito mais segura em seres humanos (Räty et al., 2008; Dormond et al., 2009). Por sua vez, é crescente a expectativa de evitar mutagênese insercional, como a observada nos ensaios para SCID-XL, por meio do desenho de vetores retrovirais ou lentivirais autoinativantes ou dotados de isoladores de cromatina, duas das mais promissoras técnicas em desenvolvimento atualmente para essa classe de vetores (Yi et al., 2005; Räty et al., 2008).
A terapia gênica para SCID-XL, por sua vez, foi curativa em 19 das 20 crianças tratadas, que apresentaram melhora significativa do seu sistema imune menos de três meses após o tratamento, bem como recuperação persistente de sua resistência a infecções (Tabela 3; Fisher & Cavazzana-Calvo, 2008; Aiuti & Roncarolo, 2009). Por seu turno, o tratamento em adolescentes não foi eficaz, sugerindo uma janela terapêutica limitada para intervenção nessa doença. Adicionam-se aos casos bem-sucedidos 30 pacientes tratados da SCID-ADA, a forma de imunodeficiência que corresponde à primeira paciente tratada por terapia gênica em 1989 (Tabela 4; Aiuti & Roncarolo, 2009).
As agências reguladoras envolvidas na autorização e no controle de ensaios clínicos em terapia gênica agiram rapidamente em ambos os casos de efeitos adversos aqui relatados. Em 1999, o ensaio que resultou na morte do paciente foi suspenso definitivamente, apesar da ausência de efeitos adversos graves nos outros 17 pacientes tratados no mesmo estudo. No caso dos ensaios para SCID-XL, os procedimentos terapêuticos já estavam encerrados, mas as autorizações para outros ensaios semelhantes foram suspensas até a avaliação criteriosa dos dados e, posteriormente, voltaram a ser concedidas. Em que pese o reconhecimento de que o procedimento de terapia gênica foi responsável pelos efeitos adversos, os comitês reguladores concluíram que nenhum desses eventos, assim como outros efeitos adversos relatados ocasionalmente, justifica a abolição de ensaios clínicos em terapia gênica. Na verdade, a análise dos efeitos adversos tem contribuído para orientar o desenvolvimento biotecnológico na área e, ao mesmo tempo, aperfeiçoar a regulamentação e os critérios para autorização de ensaios clínicos.
O balanço de efeitos adversos e benefícios em ensaios clínicos de terapia gênica indica que o curso do desenvolvimento dessa, assim como de outras terapias avançadas, tais como os tratamentos com células-tronco, será tanto mais seguro quanto mais bem fundamentado pela pesquisa básica e sujeito a regulamentação adequada para restringir a autorização de ensaios clínicos à condição de máxima segurança possível na época dos ensaios, porém sem tolher o avanço da pesquisa médica.
Terapia gênica e biotecnologia
Empresários da área de biotecnologia enxergam no sequenciamento do genoma humano oportunidades comerciais crescentes. O interesse reside, naturalmente, no fato de que a descoberta dos genes e, especialmente de mutações responsáveis, no todo ou em parte, por uma doença, pode levar ao desenvolvimento de testes diagnósticos ou medicamentos comercializáveis.
Entre outras ações, empresas começaram a investir no patenteamento de genes ou mesmo de sequências de fragmentos de DNA que ainda não tinham sequer sido associadas a genes propriamente ditos. Mais de três milhões de patentes relacionadas ao genoma foram solicitadas até hoje nos Estados Unidos. A legislação norte-americana em geral permite o patenteamento de genes, desde que isolados (e não apenas descritos como sequências de nucleotídeos) e acompanhados de evidência de utilidade, por exemplo, para desenvolvimento de testes diagnósticos. Entretanto, o patenteamento de genes é controverso. Por exemplo, as normas internas para avaliação da utilidade de descobertas relativas a genes, em vigor desde 2001 pelo Escritório de Patentes dos Estados Unidos (Uspto), foram e ainda são objeto de severas críticas, das quais o Uspto se defende com base nas leis de patentes vigentes nos Estados Unidos. Já o Instituto Nacional da Propriedade Industrial (Inpi), órgão brasileiro que concede patentes com validade nacional, informa em sua página que o patenteamento de genes naturais não é permitido no Brasil.
Fora do âmbito da controvérsia sobre patenteamento de genes, os vetores para terapia gênica, virais ou não virais, contendo genes terapêuticos, bem como suas aplicações específicas, são produtos de desenvolvimento tecnológico e, como tal, constituem objeto legítimo de patenteamento e eventual comercialização (Bobrow & Thomas, 2002). Centenas de patentes desse tipo têm sido solicitadas ao Uspto e a seus similares europeus e asiáticos. Dezenas de empresas vêm investindo em terapia gênica, a partir de tecnologias patenteadas de produção de vetores ou como parceiras de instituições de pesquisa (Tabela 5).
Em todo o mundo, as primeiras etapas de desenvolvimento de tecnologias para terapia gênica e muitos testes pré-clínicos estão ao alcance de grupos de pesquisa, institutos e universidades públicas, bem como entidades privadas financiadas com recursos públicos. Entretanto, a transferência da pesquisa de laboratório para o ensaio clínico geralmente demanda recursos que estão muito além da capacidade de financiamento público. Companhias de biotecnologia investem na realização desses ensaios em razão da existência de patentes que possam ser, eventualmente, exploradas comercialmente, assim como em todas as demais áreas de tecnologia. Até o momento, apenas um produto especificamente classificado como terapia gênica foi comercializado, mas outros quatro produtos encontram-se em fase adiantada no caminho da comercialização (Tabela 6).
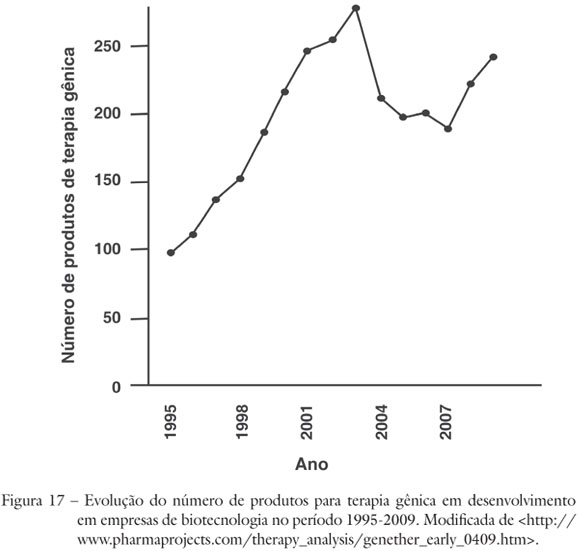
O interesse do setor industrial na terapia gênica pode ser ilustrado por dados provenientes de organizações especializadas em prospecção tecnológica. A análise da evolução do número de produtos destinados à terapia gênica, em fase de desenvolvimento por parte de empresas, revela um aspecto importante. Enquanto a produção científica na área cresce continuamente (Figura 5), a curva de crescimento do investimento industrial apresentou uma clara redução entre 2003 e 2007 (Figura 17), provavelmente influenciada pelos efeitos adversos dos ensaios de terapia gênica para SCID-XL, que foram amplamente divulgados e, naturalmente, devem ter despertado preocupação por parte dos investidores. Ainda assim, o número de produtos industriais nas fases II e III continuou crescendo no período (Figura 18) e a recuperação do crescimento do setor é previsível. No meio empresarial, há uma expectativa crescente de sucessos a um prazo compatível com os investimentos tanto na pesquisa acadêmica quanto no setor privado (Phacilitate, 2009). Um estudo estratégico de 2008 previu um mercado mundial de cerca de US$ 500 milhões de dólares em 2015 para produtos de terapia gênica (Global Industry Analysts Gene Therapy: a global strategic business report, 2008).
Terapia gênica no Brasil
Apesar da história e do reconhecimento internacional da genética brasileira, ainda há poucos grupos de pesquisa dedicados a estudos sobre terapia gênica, incluindo vacinas de DNA. Até recentemente havia pouco investimento público nessa área de investigação e nenhum interesse por parte do setor privado. O quadro, no entanto, está começando a mudar com algumas iniciativas, ainda que modestas, em ambos os setores.
A Rede de Terapia Gênica
A partir de 2005, começou a ser organizada no Brasil uma Rede de Terapia Gênica. Essa rede, coordenada pelo autor, congregou inicialmente 14 grupos de pesquisa de três Estados (Rio de Janeiro, São Paulo e Rio Grande do Sul), dedicados à pesquisa na área de terapia gênica e vacinas de DNA. Os estudos envolvem desenvolvimento de vetores virais, pesquisa básica e testes pré-clínicos nas áreas de câncer, doenças genéticas, doenças neurodegenerativas e vacinas de DNA para dengue, doença de Chagas, infecções por estreptococos e câncer.
Um primeiro ensaio clínico de terapia gênica para revascularização miocárdica com emprego de vetores plasmidiais contendo o gene do VEGF (Vascular Endothelial Growth Factor), promovido conjuntamente pelo Instituto de Cardiologia do Rio Grande do Sul, pela Fundação de Amparo à Pesquisa do Rio Grande do Sul e pela Rede de Terapia Gênica, por meio do Programa dos Institutos do Milênio do MCT/CNPq, iniciou-se em fevereiro de 2009 em Porto Alegre (http://clinicaltrials.gov/ct2/show/NCT00744315). Trata-se do primeiro ensaio clínico de terapia gênica sediado na América do Sul, em meio a dezenas de ensaios clínicos promovidos por empresas multinacionais ou instituições de pesquisa estrangeiras que contam com participação de pesquisadores sul-americanos (Tabela 7).
Terapia gênica e biotecnologia no Brasil
Consistente com a incipiente presença da pesquisa em terapia gênica, é reduzido o interesse do setor privado nessa área no país. Recentemente, no entanto, foi instalada uma empresa sediada na Fundação Parque de Alta Tecno-logia de Petrópolis, no Estado do Rio de Janeiro, que, entre outros serviços de natureza biotecnológica, está começando a oferecer suporte para ensaios de terapia gênica no país.
O vetor para o ensaio clínico de revascularização miocárdica, iniciado em Porto Alegre, foi produzido por essa empresa de serviços, um evento pioneiro no país e prenúncio de novas parcerias entre o setor privado e instituições acadêmicas nessa área de investigação científica.
Por sua vez, a consciência do papel crucial dos mecanismos de proteção à propriedade intelectual nessa área vem criando hábitos em pesquisadores antes desacostumados com a preocupação em patentear produtos e processos de interesse biotecnológico. Assim, a atuação da Rede de Terapia Gênica também estimulou o primeiro depósito internacional de patente em terapia gênica sensu stricto efetuado por uma instituição brasileira, consequente à pesquisa no laboratório do autor (World Intellectual Property Organization WO2009/121157 PCT/BR2009/000093).
Conclusão
Ainda estamos no limiar da história da terapia gênica e tudo o que se fez até hoje são os primeiros passos de uma longa e tortuosa caminhada (Flotte, 2007). Mas já há alguns sucessos pontuais que demonstram a viabilidade de incorporação da terapia gênica à prática médica. Os principais avanços, até o momento, encontram-se nas áreas de hemofilia, alguns tipos de câncer, síndromes de imunodeficiência combinada severa e certas retinopatias.
Tem havido grande progresso no planejamento e na construção de novos vetores mais seguros e eficientes (Räty et al., 2008). Em particular, as respostas imunitárias dos pacientes estão sendo estudadas em profundidade, novos modelos de estudo em animais vêm sendo desenvolvidos e a pesquisa está avançando no sentido de aumentar a segurança dos ensaios clínicos.
Os problemas não são triviais. Basta lembrar que, depois de todo o progresso da medicina até os dias de hoje, apesar do sucesso que se obteve em novos tratamentos e na prevenção de tantas doenças nos últimos 150 anos, ainda lutamos contra doenças incuráveis, que desafiam a imaginação e a competência científica e tecnológica de todo o mundo científico.
Há, no entanto, razões para otimismo e a expectativa de sucesso das tecnologias de terapia gênica vem aumentando paulatinamente. Um sinal da viabilidade de aplicação de terapia gênica em futuro próximo é o investimento crescente que empresas de biotecnologia estão fazendo no desenvolvimento e na submissão de pedidos de liberação de produtos biológicos relativos à terapia gênica.
O Brasil prepara-se para participar do advento da terapia gênica na prática médica. O contingente de cientistas, técnicos, médicos e empresários envolvidos nesse campo no país ainda é minúsculo, comparado aos países do Primeiro Mundo. Mas a decisão de investir nessa área, tanto do ponto de vista financeiro quanto do ponto de vista científico e educacional, seguramente terá retorno significativo para a medicina brasileira do século XXI.
Referências
AIUTI, A. et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med., v.360, n.5, p.447-58, 2009.
AIUTI, A.; RONCAROLO, M. G. Ten years of gene therapy for primary immune deficiencies. Hematology Am. Soc. Hematol. Educ. Program, p.682-9, 2009.
ANDERSON, W. F. Human gene therapy: the initial concepts. In: BRIGHAM, K. L. (Ed.) Gene therapy for diseases of the lung. S. l.: CRC Press, 1990. p.3-16.
ANDERSON, W. F. et al. The ADA human gene therapy clinical protocol: Points to Consider response with clinical protocol. Hum. Gene Ther., v.1, n.3, p.331-62, 1990.
ANDREWES, C. Richard Edwin Shope. In: Biographical memoirs. s. l.: National Academy of Sciences, 1979. v.50, p.352-75.
ANDREWES, C. H. Rhinoviruses and common colds. Annu. Rev. Med., v.17 p.361-70, 1966.
ATKINS, G. J. et al. Therapeutic and prophylactic applications of alphavirus vectors. Expert Rev. Mol. Med., v.10, p.e33, 2008.
BAGLEY, J. et al. Gene therapy in type 1 diabetes. Crit. Rev. Immunol., v.28, n.4, p.301-24, 2008.
BAINBRIDGE, J. W. et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N. Engl. J. Med., v.358, n.21, p.2231-9, 2008.
BAUZON, M.; HERMISTON, T. W. Exploiting diversity: genetic approaches to creating highly potent and efficacious oncolytic viruses. Curr. Opin. Mol. Ther., v.10, n.4, p.350-5, 2008.
BERETA, G. et al. Impact of retinal disease-associated RPE65 mutations on retinoid isomerization. Biochemistry, v.47, n.37, p.9856-65, 2008.
BLAESE, R. M. et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science, v.270, n.5235, p.475-80, 1995.
BOBROW, M.; THOMAS, S. Patenting DNA. Curr. Opin. Mol. Ther., v.4, n.6, p.542-7, 2002.
BRAAK, H.; DEL TREDICI, K. Assessing fetal nerve cell grafts in Parkinson's disease. Nat. Med., v.14, n.5, p.483-5, 2008.
BRINKMAN, R. R. et al. Human monogenic disorders a source of novel drug targets. Nat. Rev. Genet., v.7, n.4, p.249-60, 2006.
BUCKLEY, R. H. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu. Rev. Immunol., v.22, p.625-55, 2004.
CACABELOS, R. Molecular pathology and pharmacogenomics in Alzheimer's disease: polygenic-related effects of multifactorial treatments on cognition, anxiety and depression. Methods Find Exp. Clin. Pharmacol., v.29, Suppl A, p.1-91, 2007.
CARDONE, M. Prospects for gene therapy in inherited neurodegenerative diseases. Curr. Opin. Neurol., v.20, n.2, p.151-8, 2007.
CATTANEO, R. et al. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat. Rev. Microbiol., v.6, n.7, p.529-40, 2008.
CAVAZZANA-CALVO, M.; FISCHER, A. Gene therapy for severe combined immunodeficiency: are we there yet? J. Clin. Invest., v.117, n.6, p.1456-65, 2007.
CEMAZAR, M.; SERSA, G. Electrotransfer of therapeutic molecules into tissues. Curr. Opin. Mol. Ther., v.9, n.6, p.554-62, 2007.
CHEN, Q. et al. Gene therapy for Parkinson's disease: progress and challenges. Curr. Gene Ther., v.5, p.71-80, 2005.
CHRISTINE, C. W. et al.. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology, v.73, n.20, p.1662-9, 2009.
CIDECIYAN, A. V. et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA, v.105, n.39, p.15112-7, 2008.
CLANCHY, F. I.; WILLIAMS, R. O. Plasmid DNA as a safe gene delivery vehicle for treatment of chronic inflammatory disease. Expert Opin. Biol. Ther., v.8, n.10, p.1507-19, 2008.
COLEMAN, W. B.; TSONGALIS, G. J. (Ed.) Molecular pathology: the molecular basis of human disease. S. l.: Academic Press, 2009. 664p.
CULVER, K. W. et al. Correction of ADA deficiency in human T lymphocytes using retroviral-mediated gene transfer. Transplant Proc., v.23, n.1 Pt. 1, p.170-1, 1991.
DASS, C. R. Lipoplex-mediated delivery of nucleic acids: factors affecting in vivo transfection. J. Mol. Med., v.82, n.9, p.579-91, 2004.
DAUER, W.; PRZEDBORSKI, S. Parkinson's disease: mechanisms and models. Neuron, v.39, p.889-909, 2003.
DAWSON, T. M.; DAWSON, V. L. Molecular pathways of neurodegeneration in Parkinson's disease. Science, v.302, p.819-22, 2003.
HOLLANDER, A. I. den et al. FP Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin Eye Res., v.27, n.4, p.391-419, 2008.
DIAMOND, A.; JANKOVIC, J. The effect of deep brain stimulation on quality of life in movement disorders. J. Neurol. Neurosurg Psychiatry, v.76, p.1188-93, 2005.
DORMOND, E. et al. From the first to the third generation adenoviral vector: what parameters are governing the production yield? Biotechnol. Adv., v.27, n.2, p.133-44, 2009.
EINSTEIN, O.; BEN-HUR, T. The changing face of neural stem cell therapy in neurologic diseases. Arch. Neurol., v.65, n.4, p.452-6, 2008.
EVAN, G.; LITTLEWOOD, T. A matter of life and cell death. Science, v.281, n.5381, p.1317-22, 1998.
FAVARD, C. et al. Electrotransfer as a non viral method of gene delivery. Curr. Gene Ther., v.7, n.1, p.67-77, 2007.
FISCHER, A.; CAVAZZANA-CALVO, M Gene therapy of inherited diseases. Lancet, v.371, n.9629, p.2044-7, 2008.
FJORD-LARSEN, L. et al. Efficient in vivo protection of nigral dopaminergic neurons by lentiviral gene transfer of a modified Neurturin construct. Exp. Neurol., v.195, p.49-60, 2005.
FLOTTE, T. R. Gene therapy: the first two decades and the current state-of-the-art. J. Cell Physiol., v.213, n.2, p.301-5, 2007.
FRIEDMANN T. The road toward human gene therapy--a 25-year perspective. Ann Med., v.29 n.6 p.575-7, 1997.
FRIEDMANN, T. Stanfield Rogers: insights into virus vectors and failure of an early gene therapy model. Mol. Ther., v.4, n.4, p.285-8, 2001.
GASPAR, H. B. et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet, v.364, n.9452, p.2181-7, 2004.
GAUTHIER, S.; POIRIER, J. Current and future management of Alzheimer's disease. Alzheimers Dement., v.4, n.1, Suppl. 1, p.S48-50, 2008.
GILL, D. R. et al. Progress and prospects: the design and production of plasmid vectors. Gene Ther., v.16, n.2, p.165-71, 2009.
GOLDSTEIN, M. Decade of the brain: an agenda for the nineties. West. J. Med., v.161, p.239-41, 1994.
GREEN, D. R.; EVAN, G. I. A matter of life and death. Cancer Cell., v.1, n.1, p.19-30, 2002.
GRIBBEN, J. G. Stem cell transplantation in chronic lymphocytic leukemia. Biol. Blood Marrow Transplant., v.15, n.1, Suppl., p.53-8, 2008.
GUTTMAN, M. et al. Current concepts in the diagnosis and management of Parkinson's disease. CMAJ, v.168, p.293-301, 2003.
HACEIN-BEY-ABINA, S. et al. Gene therapy of X-linked severe combined immunodeficiency. Int. J. Hematol., v.76, n.4, p.295-8, 2002.
HACEIN-BEY-ABINA, S. et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science, v.302, n.5644, p.415-9, 2003.
HAN, J. J.; MCDONALD, C. M. Diagnosis and clinical management of spinal muscular atrophy. Phys. Med. Rehabil. Clin. N. Am., v.19, n.3, p.661-80, 2008.
HASBROUCK, N. C.; HIGH, K. A. AAV-mediated gene transfer for the treatment of hemophilia B: problems and prospects. Gene Ther., v.15, n.11, p.870-5, 2008.
HAUSER, O. et al. Encapsulated, genetically modified cells producing in vivo therapeutics. Curr. Opin. Mol. Ther., v.6, n.4, p.412-20, 2004.
HAUSWIRTH. W. et al. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene Ther., v.19, n.10, p.979-90, 2008.
HERWEIJER, H.; WOLFF, J. A. Gene therapy progress and prospects: hydrodynamic gene delivery. Gene Ther., v.14, n.2, p.99-107, 2007.
HOWE, S. J. et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin Invest., v.118, n.9, p.3143-50, 2008.
IMMONEN, A. et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol. Ther., v.10, n.5, p.967-72, 2004.
JACKSON D. A.; SYMONS R. H., BERG, P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA. v.69 n.10 p.2904-9, 1972.
JALBERT, J. J. et al. Dementia of the Alzheimer type. Epidemiol. Rev., v.30, p.15-34, 2008.
KANG, U. J. et al. Gene therapy for Parkinson's disease: determining the genes necessary for optimal dopamine replacement in rat models. Hum. Cell., v.14, n.1, p.39-48, 2001.
KAPLITT, M. G. et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet, v.369, n.9579, p.2097-105, 2007.
KOHN, D. B.; CANDOTTI, F. Gene therapy fulfilling its promise. N. Engl. J. Med., v.360, n.5, p.518-21, 2009.
KORDOWER, J. H. et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med., v.14, n.5, p.504-6, 2008.
LANDER, E. S. et al. Initial sequencing and analysis of the human genome. Nature, v.409, n.6822, p.860-921, 2001.
LI, J. Y. et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med., v.14, n.5, p.501-3, 2008.
LINDEN, R. Genes contra doenças. Terapia gênica: uma nova era na genética. Rio de Janeiro: Vieira e Lent, 2008. 128p.
LINDEN, R.; LENZ, G. Terapia gênica em neurologia. In: MORALES, M. M. (Ed.) Terapias avançadas. Rio de Janeiro: Atheneu, 2007. p.205-22.
LINDVALL, O.; WAHLBERG, L. U. Encapsulated cell biodelivery of GDNF: a novel clinical strategy for neuroprotection and neuroregeneration in Parkinson's disease? Exp. Neurol., v.209, n.1, p.82-8, 2008.
LUNDBERG, C. et al. Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr. Gene Ther., v.8, n.6, p.461-73, 2008.
LUO J. et al. Subthalamic GAD gene therapy in a Parkinson's disease rat model. Science, v.298, p.425-9, 2002.
MACHIDA, C. A. (Ed.) Viral vectors for gene therapy: methods and protocols. S. l.: Humana Press, 2002. 608p.
MAGUIRE, A. M. et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N. Engl. J. Med., v.358, n.21, p.2240-8, 2008.
MANNO, C. S. et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood, v.101, n.8, p.2963-72, 2003.
MANNO, C. S. et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med., v.12, n.3, p.342-7, 2006.
MARKS, W. J. et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson's disease: an open-label, phase I trial. Lancet Neurol., v.7, n.5, p.400-8, 2008.
MENDEZ, I. et al. Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nat. Med., v.14, n.5, p.507-9, 2008.
MERCOLA, K. E.; CLINE, M. J. Sounding boards. The potentials of inserting new genetic information. N. Engl. J. Med., v.303, n.22, p.1297-300, 1980.
MINGOZZI, F.; HIGH, K. A. Immune responses to AAV in clinical trials. Curr. Gene Ther., v.7, n.5, p.316-24, 2007.
MULLEN C. A. et al. Molecular analysis of T lymphocyte-directed gene therapy for adenosine deaminase deficiency: long-term expression in vivo of genes introduced with a retroviral vector. Hum. Gene Ther., v.7 n.9 p.1123-9, 1996.
MUUL, L. M. et al. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood, v.101, n.7, p.2563-9, 2003.
NAKANO, K. Neural circuits and topographic organization of the basal ganglia and related regions. Brain Dev., v.22, Suppl. 1, p.S5-16, 2000.
NATHWANI, A. C. et al. A review of gene therapy for haematological disorders. Br. J. Haematol., v.28, n.1, p.3-17, 2005.
NCBI. Genes and disease. 2009. Disponível em: <http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=gnd&ref=sidebar> .
O'CONNOR, T. P.; CRYSTAL, R. G. Genetic medicines: treatment strategies for hereditary disorders. Nat. Rev. Genet., v.7, n.4, p.261-76, 2006.
OLANOW, C. W. et al. Why have we failed to achieve neuroprotection in Parkinson's disease? Ann Neurol., v.64 Suppl 2,p.S101-10, 2008.
PARDRIDGE, W. M. Tyrosine hydroxylase replacement in experimental Parkinson's disease with transvascular gene therapy. NeuroRx., v.2, n.1, p.129-38, 2005.
PARDRIDGE, W. M. Blood-brain barrier delivery of protein and non-viral gene therapeutics with molecular Trojan horses. J. Control. Release, v.122, n.3, p.345-8, 2007.
PEARSON, S. et al. China approves first gene therapy. Nature Biotechnol., v.22, p.3-4, 2004.
PHACILITATE, Inc. 5th Annual Cell and Gene Therapy Forum. Washington, DC, 26-28 January, 2009.
POEHNER, W. J. et al. A homozygous deletion in RPE65 in a small Sardinian family with autosomal recessive retinal dystrophy. Mol. Vis., v.6, p.192-8, 2000.
POEWE, W. Treatments for Parkinson disease - past achievements and current clinical needs. Neurology, v.72, n.7, Supp. p.S65-73, 2009.
PORTEUS, M. H. et al. A look to future directions in gene therapy research for monogenic diseases. PLoS Genet., v.2, n.9, p.e133, 2006.
PROCHOWNIK, E. V. c-Myc: linking transformation and genomic instability. Curr. Mol. Med., v.8, n.6, p.446-58, 2008.
RADUNOVIC, A. et al. Clinical care of patients with amyotrophic lateral sclerosis. Lancet Neurol., v.6, n.10, p.913-25, 2007.
RAINOV, N. G.; REN, H. Gene therapy for human malignant brain tumors. Cancer J., v.9, n.3, p.180-8, 2003.
RAPER, S. E. et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab., v.80, n.1-2, p.148-58, 2003.
RAPER, S. E. et al. Developing adenoviral-mediated in vivo gene therapy for ornithine transcarbamylase deficiency. J. Inherit Metab. Dis., v.21, Suppl. 1, p.119-37, 1998.
RÄTY, J. K. et al. Improving safety of gene therapy. Curr. Drug Saf., v.3, n.1, p.46-53, 2008.
REFFELMANN, T. et al. Promise of blood- and bone marrow-derived stem cell transplantation for functional cardiac repair: putting it in perspective with existing therapy. J. Am. Coll. Cardiol., v.53, n.4, p.305-8, 2008.
RIBACKA, C. et al. Cancer, stem cells, and oncolytic viruses. Ann. Med., v.40, n.7, p.496-505, 2008.
ROGERS, S.; ROUS, P. Joint action of a chemical carcinogen and a neoplastic virus to induce cancer in rabbits; results of exposing epidermal cells to a carcinogenic hydrocarbon at time of infection with the Shope papilloma virus. J. Exp. Med., v.93, n.5, p.459-88, 1951.
ROGERS, S. Serial transplantation of rabbit papillomas caused by the Shope virus. J. Exp. Med., v.95, n.6, p.543-54, 1952.
SANVICENS, N.; MARCO, M. P. Multifunctional nanoparticles--properties and prospects for their use in human medicine. Trends Biotechnol., v.26, n.8, p.425-33, 2008.
SCAGLIA, F.; LEE, B. Clinical, biochemical, and molecular spectrum of hyperargininemia due to arginase I deficiency. Am. J. Med. Genet. C. Semin. Med. Genet., v.142C, n.2, p.113-20, 2006.
SILVA, C. L. et al. Recent advances in DNA vaccines for autoimmune diseases. Expert Rev. Vaccines, v.8, n.2, p.239-52, 2009.
SYKES, K. Progress in the development of genetic immunization. Expert Rev. Vaccines, v.7, n.9, p.1395-404, 2008.
THAJEB, P. et al. The effects of storage conditions and trophic supplementation on the survival of fetal mesencephalic cells. Cell Transplant., v.6, p.297-307, 1997.
TORRENTE, Y.; POLLI, E. Mesenchymal stem cell transplantation for neurodegenerative diseases. Cell Transplant., v.17, n.10-11, p.1103-13, 2008.
Van der SPUY, J. et al. Predominant rod photoreceptor degeneration in Leber congenital amaurosis. Mol Vis., v.11, p.542-53, 2005.
VAZQUEZ, A. et al. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov., v.7, n.12, p.979-87, 2008.
VELLAI, T.; VIDA, G. The origin of eukaryotes: the difference between prokaryotic and eukaryotic cells. Proc. Biol. Sci., v.266, n.1428, p.1571-7, 1999.
VENTER, J. C. et al. The sequence of the human genome. Science, v.291, n.5507, p.1304-51, 2001.
VOSS, C. Production of plasmid DNA for pharmaceutical use. Biotechnol. Annu. Rev., v.13, p.201-22, 2007.
WANG, X. et al. Mitotic checkpoint defects in human cancers and their implications to chemotherapy. Front Biosci., v.13, p.2103-14, 2008.
WATSON, J. D. et al. Recombinant DNA: genes and genomics: a short course. S. l.: Freeman, 2006. 474p.
WU, C.; LU, Y. Inclusion of high molecular weight dextran in calcium phosphate-mediated transfection significantly improves gene transfer efficiency. Cell. Mol. Biol., Noisy-le-grand, v.53, n.4, p.67-74, 2007.
YI, Y.; HAHM, S. H.; LEE, K. H. Retroviral gene therapy: safety issues and possible solutions. Curr. Gene Ther., v.5 n.1 p.25-35, 2005.
Recebido em 29.7.2010. Aceito em 17.8.2010.
Rafael Linden é médico, doutor em Ciências, professor titular do Instituto de Bio-física Carlos Chagas Filho, UFRJ. @ rlinden@biof.ufrj.br
- AIUTI, A. et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med, v.360, n.5, p.447-58, 2009.
- AIUTI, A.; RONCAROLO, M. G. Ten years of gene therapy for primary immune deficiencies. Hematology Am. Soc. Hematol. Educ. Program, p.682-9, 2009.
- ANDERSON, W. F. Human gene therapy: the initial concepts. In: BRIGHAM, K. L. (Ed.) Gene therapy for diseases of the lung S. l.: CRC Press, 1990. p.3-16.
- ANDERSON, W. F. et al. The ADA human gene therapy clinical protocol: Points to Consider response with clinical protocol. Hum. Gene Ther, v.1, n.3, p.331-62, 1990.
- ANDREWES, C. Richard Edwin Shope. In: Biographical memoirs s. l.: National Academy of Sciences, 1979. v.50, p.352-75.
- BAGLEY, J. et al. Gene therapy in type 1 diabetes. Crit. Rev. Immunol, v.28, n.4, p.301-24, 2008.
- BAINBRIDGE, J. W. et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N. Engl. J. Med, v.358, n.21, p.2231-9, 2008.
- BERETA, G. et al. Impact of retinal disease-associated RPE65 mutations on retinoid isomerization. Biochemistry, v.47, n.37, p.9856-65, 2008.
- BLAESE, R. M. et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science, v.270, n.5235, p.475-80, 1995.
- BOBROW, M.; THOMAS, S. Patenting DNA. Curr. Opin. Mol. Ther, v.4, n.6, p.542-7, 2002.
- BRAAK, H.; DEL TREDICI, K. Assessing fetal nerve cell grafts in Parkinson's disease. Nat. Med, v.14, n.5, p.483-5, 2008.
- BRINKMAN, R. R. et al. Human monogenic disorders a source of novel drug targets. Nat. Rev. Genet, v.7, n.4, p.249-60, 2006.
- BUCKLEY, R. H. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu. Rev. Immunol, v.22, p.625-55, 2004.
- CACABELOS, R. Molecular pathology and pharmacogenomics in Alzheimer's disease: polygenic-related effects of multifactorial treatments on cognition, anxiety and depression. Methods Find Exp. Clin. Pharmacol, v.29, Suppl A, p.1-91, 2007.
- CARDONE, M. Prospects for gene therapy in inherited neurodegenerative diseases. Curr. Opin. Neurol, v.20, n.2, p.151-8, 2007.
- CAVAZZANA-CALVO, M.; FISCHER, A. Gene therapy for severe combined immunodeficiency: are we there yet? J. Clin. Invest., v.117, n.6, p.1456-65, 2007.
- CEMAZAR, M.; SERSA, G. Electrotransfer of therapeutic molecules into tissues. Curr. Opin. Mol. Ther, v.9, n.6, p.554-62, 2007.
- CHEN, Q. et al. Gene therapy for Parkinson's disease: progress and challenges. Curr. Gene Ther, v.5, p.71-80, 2005.
- CHRISTINE, C. W. et al.. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology, v.73, n.20, p.1662-9, 2009.
- CIDECIYAN, A. V. et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA, v.105, n.39, p.15112-7, 2008.
- CLANCHY, F. I.; WILLIAMS, R. O. Plasmid DNA as a safe gene delivery vehicle for treatment of chronic inflammatory disease. Expert Opin. Biol. Ther, v.8, n.10, p.1507-19, 2008.
- COLEMAN, W. B.; TSONGALIS, G. J. (Ed.) Molecular pathology: the molecular basis of human disease. S. l.: Academic Press, 2009. 664p.
- CULVER, K. W. et al. Correction of ADA deficiency in human T lymphocytes using retroviral-mediated gene transfer. Transplant Proc, v.23, n.1 Pt. 1, p.170-1, 1991.
- DASS, C. R. Lipoplex-mediated delivery of nucleic acids: factors affecting in vivo transfection. J. Mol. Med, v.82, n.9, p.579-91, 2004.
- DAUER, W.; PRZEDBORSKI, S. Parkinson's disease: mechanisms and models. Neuron, v.39, p.889-909, 2003.
- DAWSON, T. M.; DAWSON, V. L. Molecular pathways of neurodegeneration in Parkinson's disease. Science, v.302, p.819-22, 2003.
- HOLLANDER, A. I. den et al. FP Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin Eye Res, v.27, n.4, p.391-419, 2008.
- DIAMOND, A.; JANKOVIC, J. The effect of deep brain stimulation on quality of life in movement disorders. J. Neurol. Neurosurg Psychiatry, v.76, p.1188-93, 2005.
- DORMOND, E. et al. From the first to the third generation adenoviral vector: what parameters are governing the production yield? Biotechnol. Adv., v.27, n.2, p.133-44, 2009.
- EINSTEIN, O.; BEN-HUR, T. The changing face of neural stem cell therapy in neurologic diseases. Arch. Neurol., v.65, n.4, p.452-6, 2008.
- EVAN, G.; LITTLEWOOD, T. A matter of life and cell death. Science, v.281, n.5381, p.1317-22, 1998.
- FAVARD, C. et al. Electrotransfer as a non viral method of gene delivery. Curr. Gene Ther, v.7, n.1, p.67-77, 2007.
- FISCHER, A.; CAVAZZANA-CALVO, M Gene therapy of inherited diseases. Lancet, v.371, n.9629, p.2044-7, 2008.
- FJORD-LARSEN, L. et al. Efficient in vivo protection of nigral dopaminergic neurons by lentiviral gene transfer of a modified Neurturin construct. Exp. Neurol, v.195, p.49-60, 2005.
- FLOTTE, T. R. Gene therapy: the first two decades and the current state-of-the-art. J. Cell Physiol, v.213, n.2, p.301-5, 2007.
- FRIEDMANN T. The road toward human gene therapy--a 25-year perspective. Ann Med, v.29 n.6 p.575-7, 1997.
- FRIEDMANN, T. Stanfield Rogers: insights into virus vectors and failure of an early gene therapy model. Mol. Ther., v.4, n.4, p.285-8, 2001.
- GASPAR, H. B. et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet, v.364, n.9452, p.2181-7, 2004.
- GAUTHIER, S.; POIRIER, J. Current and future management of Alzheimer's disease. Alzheimers Dement, v.4, n.1, Suppl. 1, p.S48-50, 2008.
- GILL, D. R. et al. Progress and prospects: the design and production of plasmid vectors. Gene Ther, v.16, n.2, p.165-71, 2009.
- GOLDSTEIN, M. Decade of the brain: an agenda for the nineties. West. J. Med, v.161, p.239-41, 1994.
- GREEN, D. R.; EVAN, G. I. A matter of life and death. Cancer Cell, v.1, n.1, p.19-30, 2002.
- GRIBBEN, J. G. Stem cell transplantation in chronic lymphocytic leukemia. Biol. Blood Marrow Transplant, v.15, n.1, Suppl., p.53-8, 2008.
- GUTTMAN, M. et al. Current concepts in the diagnosis and management of Parkinson's disease. CMAJ, v.168, p.293-301, 2003.
- HACEIN-BEY-ABINA, S. et al. Gene therapy of X-linked severe combined immunodeficiency. Int. J. Hematol, v.76, n.4, p.295-8, 2002.
- HACEIN-BEY-ABINA, S. et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science, v.302, n.5644, p.415-9, 2003.
- HAN, J. J.; MCDONALD, C. M. Diagnosis and clinical management of spinal muscular atrophy. Phys. Med. Rehabil. Clin. N. Am, v.19, n.3, p.661-80, 2008.
- HASBROUCK, N. C.; HIGH, K. A. AAV-mediated gene transfer for the treatment of hemophilia B: problems and prospects. Gene Ther, v.15, n.11, p.870-5, 2008.
- HAUSER, O. et al. Encapsulated, genetically modified cells producing in vivo therapeutics. Curr. Opin. Mol. Ther, v.6, n.4, p.412-20, 2004.
- HAUSWIRTH. W. et al. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene Ther, v.19, n.10, p.979-90, 2008.
- HERWEIJER, H.; WOLFF, J. A. Gene therapy progress and prospects: hydrodynamic gene delivery. Gene Ther, v.14, n.2, p.99-107, 2007.
- HOWE, S. J. et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin Invest, v.118, n.9, p.3143-50, 2008.
- IMMONEN, A. et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol. Ther., v.10, n.5, p.967-72, 2004.
- JACKSON D. A.; SYMONS R. H., BERG, P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA. v.69 n.10 p.2904-9, 1972.
- JALBERT, J. J. et al. Dementia of the Alzheimer type. Epidemiol. Rev, v.30, p.15-34, 2008.
- KANG, U. J. et al. Gene therapy for Parkinson's disease: determining the genes necessary for optimal dopamine replacement in rat models. Hum. Cell., v.14, n.1, p.39-48, 2001.
- KAPLITT, M. G. et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet, v.369, n.9579, p.2097-105, 2007.
- KOHN, D. B.; CANDOTTI, F. Gene therapy fulfilling its promise. N. Engl. J. Med., v.360, n.5, p.518-21, 2009.
- KORDOWER, J. H. et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med, v.14, n.5, p.504-6, 2008.
- LANDER, E. S. et al. Initial sequencing and analysis of the human genome. Nature, v.409, n.6822, p.860-921, 2001.
- LI, J. Y. et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med., v.14, n.5, p.501-3, 2008.
- LINDEN, R. Genes contra doenças Terapia gênica: uma nova era na genética. Rio de Janeiro: Vieira e Lent, 2008. 128p.
- LINDEN, R.; LENZ, G. Terapia gênica em neurologia. In: MORALES, M. M. (Ed.) Terapias avançadas Rio de Janeiro: Atheneu, 2007. p.205-22.
- LINDVALL, O.; WAHLBERG, L. U. Encapsulated cell biodelivery of GDNF: a novel clinical strategy for neuroprotection and neuroregeneration in Parkinson's disease? Exp. Neurol, v.209, n.1, p.82-8, 2008.
- LUNDBERG, C. et al. Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr. Gene Ther, v.8, n.6, p.461-73, 2008.
- LUO J. et al. Subthalamic GAD gene therapy in a Parkinson's disease rat model. Science, v.298, p.425-9, 2002.
- MACHIDA, C. A. (Ed.) Viral vectors for gene therapy: methods and protocols. S. l.: Humana Press, 2002. 608p.
- MAGUIRE, A. M. et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N. Engl. J. Med, v.358, n.21, p.2240-8, 2008.
- MANNO, C. S. et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood, v.101, n.8, p.2963-72, 2003.
- MANNO, C. S. et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med, v.12, n.3, p.342-7, 2006.
- MARKS, W. J. et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson's disease: an open-label, phase I trial. Lancet Neurol, v.7, n.5, p.400-8, 2008.
- MENDEZ, I. et al. Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nat. Med., v.14, n.5, p.507-9, 2008.
- MERCOLA, K. E.; CLINE, M. J. Sounding boards. The potentials of inserting new genetic information. N. Engl. J. Med, v.303, n.22, p.1297-300, 1980.
- MINGOZZI, F.; HIGH, K. A. Immune responses to AAV in clinical trials. Curr. Gene Ther, v.7, n.5, p.316-24, 2007.
- MULLEN C. A. et al. Molecular analysis of T lymphocyte-directed gene therapy for adenosine deaminase deficiency: long-term expression in vivo of genes introduced with a retroviral vector. Hum. Gene Ther., v.7 n.9 p.1123-9, 1996.
- MUUL, L. M. et al. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood, v.101, n.7, p.2563-9, 2003.
- NAKANO, K. Neural circuits and topographic organization of the basal ganglia and related regions. Brain Dev, v.22, Suppl. 1, p.S5-16, 2000.
- NATHWANI, A. C. et al. A review of gene therapy for haematological disorders. Br. J. Haematol., v.28, n.1, p.3-17, 2005.
- NCBI. Genes and disease. 2009. Disponível em: <http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=gnd&ref=sidebar>
- O'CONNOR, T. P.; CRYSTAL, R. G. Genetic medicines: treatment strategies for hereditary disorders. Nat. Rev. Genet, v.7, n.4, p.261-76, 2006.
- OLANOW, C. W. et al. Why have we failed to achieve neuroprotection in Parkinson's disease? Ann Neurol., v.64 Suppl 2,p.S101-10, 2008.
- PARDRIDGE, W. M. Tyrosine hydroxylase replacement in experimental Parkinson's disease with transvascular gene therapy. NeuroRx, v.2, n.1, p.129-38, 2005.
- PARDRIDGE, W. M. Blood-brain barrier delivery of protein and non-viral gene therapeutics with molecular Trojan horses. J. Control. Release, v.122, n.3, p.345-8, 2007.
- PEARSON, S. et al. China approves first gene therapy. Nature Biotechnol, v.22, p.3-4, 2004.
- PHACILITATE, Inc. 5th Annual Cell and Gene Therapy Forum. Washington, DC, 26-28 January, 2009.
- POEHNER, W. J. et al. A homozygous deletion in RPE65 in a small Sardinian family with autosomal recessive retinal dystrophy. Mol. Vis, v.6, p.192-8, 2000.
- POEWE, W. Treatments for Parkinson disease - past achievements and current clinical needs. Neurology, v.72, n.7, Supp. p.S65-73, 2009.
- PORTEUS, M. H. et al. A look to future directions in gene therapy research for monogenic diseases. PLoS Genet, v.2, n.9, p.e133, 2006.
- PROCHOWNIK, E. V. c-Myc: linking transformation and genomic instability. Curr. Mol. Med, v.8, n.6, p.446-58, 2008.
- RADUNOVIC, A. et al. Clinical care of patients with amyotrophic lateral sclerosis. Lancet Neurol, v.6, n.10, p.913-25, 2007.
- RAINOV, N. G.; REN, H. Gene therapy for human malignant brain tumors. Cancer J., v.9, n.3, p.180-8, 2003.
- RAPER, S. E. et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab, v.80, n.1-2, p.148-58, 2003.
- RAPER, S. E. et al. Developing adenoviral-mediated in vivo gene therapy for ornithine transcarbamylase deficiency. J. Inherit Metab. Dis, v.21, Suppl. 1, p.119-37, 1998.
- RÄTY, J. K. et al. Improving safety of gene therapy. Curr. Drug Saf, v.3, n.1, p.46-53, 2008.
- REFFELMANN, T. et al. Promise of blood- and bone marrow-derived stem cell transplantation for functional cardiac repair: putting it in perspective with existing therapy. J. Am. Coll. Cardiol, v.53, n.4, p.305-8, 2008.
- RIBACKA, C. et al. Cancer, stem cells, and oncolytic viruses. Ann. Med, v.40, n.7, p.496-505, 2008.
- ROGERS, S.; ROUS, P. Joint action of a chemical carcinogen and a neoplastic virus to induce cancer in rabbits; results of exposing epidermal cells to a carcinogenic hydrocarbon at time of infection with the Shope papilloma virus. J. Exp. Med, v.93, n.5, p.459-88, 1951.
- ROGERS, S. Serial transplantation of rabbit papillomas caused by the Shope virus. J. Exp. Med, v.95, n.6, p.543-54, 1952.
- SANVICENS, N.; MARCO, M. P. Multifunctional nanoparticles--properties and prospects for their use in human medicine. Trends Biotechnol, v.26, n.8, p.425-33, 2008.
- SCAGLIA, F.; LEE, B. Clinical, biochemical, and molecular spectrum of hyperargininemia due to arginase I deficiency. Am. J. Med. Genet. C. Semin. Med. Genet, v.142C, n.2, p.113-20, 2006.
- SILVA, C. L. et al. Recent advances in DNA vaccines for autoimmune diseases. Expert Rev. Vaccines, v.8, n.2, p.239-52, 2009.
- SYKES, K. Progress in the development of genetic immunization. Expert Rev. Vaccines, v.7, n.9, p.1395-404, 2008.
- THAJEB, P. et al. The effects of storage conditions and trophic supplementation on the survival of fetal mesencephalic cells. Cell Transplant., v.6, p.297-307, 1997.
- TORRENTE, Y.; POLLI, E. Mesenchymal stem cell transplantation for neurodegenerative diseases. Cell Transplant, v.17, n.10-11, p.1103-13, 2008.
- Van der SPUY, J. et al. Predominant rod photoreceptor degeneration in Leber congenital amaurosis. Mol Vis, v.11, p.542-53, 2005.
- VAZQUEZ, A. et al. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov, v.7, n.12, p.979-87, 2008.
- VELLAI, T.; VIDA, G. The origin of eukaryotes: the difference between prokaryotic and eukaryotic cells. Proc. Biol. Sci, v.266, n.1428, p.1571-7, 1999.
- VENTER, J. C. et al. The sequence of the human genome. Science, v.291, n.5507, p.1304-51, 2001.
- VOSS, C. Production of plasmid DNA for pharmaceutical use. Biotechnol. Annu. Rev, v.13, p.201-22, 2007.
- WANG, X. et al. Mitotic checkpoint defects in human cancers and their implications to chemotherapy. Front Biosci, v.13, p.2103-14, 2008.
- WATSON, J. D. et al. Recombinant DNA: genes and genomics: a short course S. l.: Freeman, 2006. 474p.
- WU, C.; LU, Y. Inclusion of high molecular weight dextran in calcium phosphate-mediated transfection significantly improves gene transfer efficiency. Cell. Mol. Biol., Noisy-le-grand, v.53, n.4, p.67-74, 2007.
- YI, Y.; HAHM, S. H.; LEE, K. H. Retroviral gene therapy: safety issues and possible solutions. Curr. Gene Ther., v.5 n.1 p.25-35, 2005.
Datas de Publicação
-
Publicação nesta coleção
23 Nov 2010 -
Data do Fascículo
2010
Histórico
-
Recebido
29 Jul 2010 -
Aceito
17 Ago 2010