Resumos
O artigo apresenta uma breve revisão sobre os achados históricos, epidemiológicos, tratamento e perspectivas terapêuticas para as epilepsias, com enfoque na epilepsia do lobo temporal. Apresenta dados obtidos com estudos de proteômica empregando tecido epiléptico e destaca a importância da aplicação desse método na busca de novos alvos terapêuticos.
Epilepsia do lobo temporal; Hipocampo; Pilocarpina; Proteômica
The article presents a brief review of the historical findings, epidemiological, and therapeutic treatment for epilepsy, with a focus on temporal lobe epilepsy. Presents data from proteomic studies in epileptic brain tissue and highlights the importance of the application of this method in the search for new therapeutic targets for temporal lobe epilepsy.
Temporal Lobe Epilepsy; Hippocampus; Pilocarpine; Proteomic
NEUROCIÊNCIAS
Epilepsia do lobo temporal: mecanismos e perspectivas
Maria José da silva Fernandes
Professora do Departamento de Neurologia e Neurocirurgia, Disciplina de Neurologia Experimental, da Universidade Federal de São Paulo(Unifesp). Membro da Comissão de Neurobiologia, "Task Force" da Liga Internacional de Epilepsia (ILAE). @ - fernandesepm@gmail.com / mjsfernandes19@unifesp.br
RESUMO
O artigo apresenta uma breve revisão sobre os achados históricos, epidemiológicos, tratamento e perspectivas terapêuticas para as epilepsias, com enfoque na epilepsia do lobo temporal. Apresenta dados obtidos com estudos de proteômica empregando tecido epiléptico e destaca a importância da aplicação desse método na busca de novos alvos terapêuticos.
Palavras-chave: Epilepsia do lobo temporal, Hipocampo, Pilocarpina, Proteômica.
ABSTRACT
The article presents a brief review of the historical findings, epidemiological, and therapeutic treatment for epilepsy, with a focus on temporal lobe epilepsy. Presents data from proteomic studies in epileptic brain tissue and highlights the importance of the application of this method in the search for new therapeutic targets for temporal lobe epilepsy.
Keywords: Temporal Lobe Epilepsy, Hippocampus, Pilocarpine, Proteomic.
Epilepsia: um breve histórico
Relatos de epilepsia datam de 2000 a.C. em textos de origem babilônica. No entanto, o termo "epilepsia" foi referido pela primeira vez na Grécia antiga, com o significado de "ser tomado, atacado, possuído", provavelmente fazendo acepção ao que ocorre durante uma crise epiléptica. Nos primórdios, a epilepsia era então considerada como fruto de possessão por entidades espirituais, e a falta de conhecimento levou diversos povos como gregos, romanos, hebreus, árabes e outros a criarem estigmas e crenças associando a epilepsia ao misticismo, tendo perdurado ao longo dos tempos, com vestígios ainda nos dias atuais.
Nem mesmo os relatos de Hipócrates (400 a.C.) e Galeno (175 d.C.) deduzindo que a epilepsia tivesse origem em anomalias decorrentes do cérebro foram capazes de alterar o comportamento ou pensamento popular daquela época, e durante a Idade Média, a Santa Inquisição perseguiu e condenou à morte muitos epilépticos considerados loucos e hereges.
Somente no século XIX, com avanços no conhecimento da neurofisiologia, é que a epilepsia passou a ser encarada pela comunidade científica como uma doença com base cerebral. Um dos pioneiros nessa área foi John Hughlings Jackson, um neurologista britânico, que propôs uma base anatômica e fisiológica organizada para a hierarquia e localização das funções cerebrais.
A partir do século XX, os avanços neurofisiológicos tornaram cada vez mais claros e consensuais aos cientistas que a epilepsia tinha origem cerebral e devia ser encarada como uma doença a ser tratada. A descoberta do neurônio e dos mecanismos de transmissão neuronal ocorrida nos séculos XVIII e XIX foi fundamental para que a epilepsia pudesse ser compreendida (Moreira, 2004). Os avanços biotecnológicos mais recentes contribuíram para o crescente conhecimento sobre os mecanismos envolvidos com a geração de crises e um dos desafios para os pesquisadores é fazer que essas informações cheguem ao maior número de pessoas possível para que os pacientes deixem de ser discriminados ou estigmatizados.
Mas o que é epilepsia?
Epilepsia é um distúrbio cerebral causado pela predisposição permanente do cérebro em gerar crises epilépticas espontâneas, recorrentes, acompanhadas de consequências neurobiológicas, cognitivas e sociais (Fisher et al, 2005).
As crises epilépticas são definidas como manifestações clínicas que refletem disfunção temporária de um conjunto de neurônios. Dependendo da localização, as crises podem ser focais, ou seja, com início em uma região restrita do encéfalo, ou generalizada, quando as descargas se originam concomitantemente nos dois hemisférios. As crises focais podem ser simples, quando há preservação da consciência durante o ictus (crise epiléptica), ou complexas, quando há perda de consciência.
As crises devem ser avaliadas juntamente com outros dados do paciente, como idade, análise de imagem e eletroencefalograma, exame físico para definir o diagnóstico sindrômico fundamental para elaboração de uma programação terapêutica (Yacubian, 2002). Quanto mais adequada for a resposta do paciente ao tratamento, melhor será o prognóstico.
Síndromes epilépticas idiopáticas são aquelas sem substrato lesional detectável por exames de imagem, porém com provável predisposição genética. As crises geralmente são bem controladas por tratamentos. As síndromes epilépticas sintomáticas são aquelas cujas crises são decorrentes de lesão estrutural em áreas do sistema nervoso central, e as síndromes provavelmente sintomáticas são aquelas em que a lesão presumida não pode ser demonstrada com exames disponíveis (Yacubian, 2002).
A epilepsia é o transtorno mais comum na clínica neurológica. Cerca de cinquenta milhões de pessoas sofrem de epilepsia com crises ativas necessitando de tratamentos, e 30% desses pacientes apresentam crises refratárias (sem resposta) ao tratamento medicamentoso disponível no mercado (WHO, 2010). Aproximadamente 90% dos casos de epilepsia ocorrem em países em desenvolvimento. Na Índia, por exemplo, estima-se um custo de 0,5% do PIB para tratar pacientes com epilepsia (~5 milhões); na Europa esse valor é de 20 bilhões de Euros (WHO, 2010). No Brasil, embora os estudos epidemiológicos sejam escassos, estima-se que a epilepsia atinja 2% a 4% da população, correspondendo a aproximadamente três milhões de pessoas em diferentes idades e classes sociais (Marchetti et al., 2005).
Além dos fatores citados como alta incidência e alto custo para tratamento dos pacientes, fatores sociais e aumento de duas a três vezes no risco de morte em pacientes com epilepsia indicam a urgência de se adotar medidas preventivas contra a epilepsia.
Quais as principais causas da epilepsia?
Um grande número de doenças ou lesão cerebral pode causar epilepsia, as causas mais frequentes são: anomalia cerebral durante o desenvolvimento; traumatismo craniano; hemorragias; anóxia durante o parto; tumores; infecção cerebral; crises prolongadas e crises febris.
A Epilepsia do Lobo Temporal Mesial (ELTM) é um tipo de epilepsia focal que apresenta grande relevância clínica em razão de alta incidência e gravidade. Pacientes com ELTM apresentam crises focais simples ou complexas que se originam em estruturas mesiais do lobo temporal, geralmente precedidas por auras. Auras correspondem a manifestações sensitivo-sensoriais, vegetativas ou psíquicas puramente subjetivas e que ocorrem em 20% a 90% dos pacientes com epilepsia do lobo temporal (Kotagal, 1991). Em geral são de curta duração, não excedendo 1 ou 2 minutos. São exemplos: aura epigástrica (sensação de náusea, mal-estar); medo; depressão e angústia (frequentemente ocorre no período interictal mas pode ocorrer antes da crise); déjà-vu, jamais vu; e pensamento forçado (impressão de estar vivenciando algo que já aconteceu antes - déjà-vu, ou que parece totalmente estranho - jamais vu, ou pensamento que ocorre repetidamente mesmo quando se tenta concentrar em outro fato); alucinações gustativas, olfativas, auditivas, somestésicas e visuais.
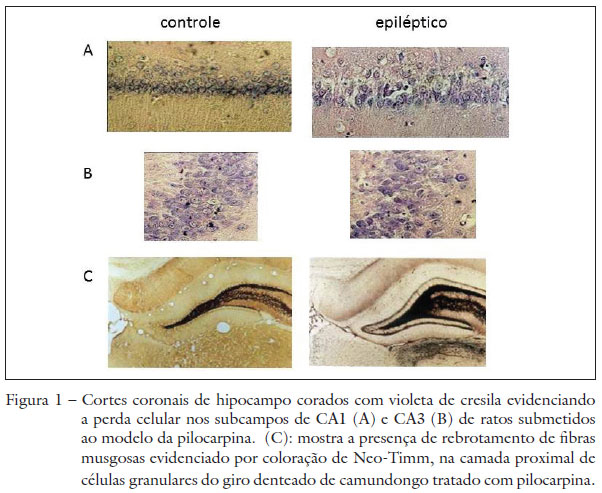
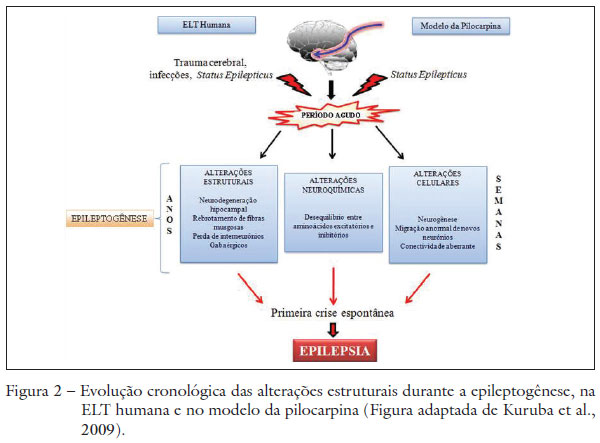
A Esclerose Hipocampal (EH) é frequentemente associada ao quadro de ELTM e é caracterizada por extensa perda celular nos subcampos CA1, CA3 e hilo da formação hipocampal, gliose e dispersão de células granulares no giro dentea- do (Babb, 1987). A esclerose hipocampal é parte de um conjunto de alterações que ocorrem durante a epileptogênese causada a partir de um insulto ao SNC. O termo epileptogênese refere-se ao processo dinâmico que progressivamente altera a excitabilidade neuronal, estabelece conexões críticas e induz alterações estruturais. Além da esclerose hipocampal, a epilepsia é acompanhada por neurogênese, rebrotamento de fibras musgosas (crescimento exacerbado de axônios de células granulares do giro denteado em decorrência da perda de seus alvos, as células musgosas, criando uma alça de autoestimulação pelas células granulares com aumento de excitabilidade), dano à barreira hematoencefálica, recrutamento de células inflamatórias pelo cérebro, reorganização da matriz extracelular e reorganização da citoarquitetura de neurônios (Sutula et al., 2004; Babb et al., 1991; Pitkanen, 2010). Um dado recente na literatura é que essas alterações desencadeadas por um insulto inicial podem continuar a progredir mesmo após a epilepsia ser diagnosticada, ou seja, o processo é progressivo. Daí a importância de se conhecer que fatores são alterados nas diferentes fases da epileptogênese.
O uso de modelos experimentais é bastante apropriado para buscar essas informações.
O que é modelo experimental de epilepsia?
Denominamos modelo experimental de epilepsia a possibilidade de reproduzir, ou modelar, características da epilepsia da qual temos interesse de estudar. Várias espécies de roedores (rato, camundongo, cobaia, Proechymis e gerbilo), insetos (drosófila), primatas (Papio, macaca, saguis), aves (Gallus) etc. já foram empregados em estudos para modelar epilepsia. No entanto, os mais utilizados são camundongos e ratos pelo baixo custo, praticidade de se criar em laboratório e pelo conhecimento adquirido quanto às bases neuroanatômicas, neuroquímicas e comportamentais. Os modelos experimentais de epilepsia podem ser obtidos por meio de preparações in vivo, quando o animal é submetido a um protocolo de indução de crises por agente químico, físico (criogenia) ou elétrico, e é mantido vivo para acompanhamento da epileptogênese, e in vitro, quando se empregam fatias de tecido, cultura de células ou células dispersas para estudar substâncias ou procedimentos convulsivantes (para detalhes, ver Loscher, 2011).
Um dos modelos que têm sido muito utilizados para modelar a epilepsia sintomática (adquirida) com lesão identificável no SNC é o modelo de epilepsia induzido por pilocarpina (Cavalheiro, 1995). A pilocarpina, um agonista colinérgico muscarínico, quando administrado por via sistêmica em altas doses em roedores (~360 mg/kg), induz um quadro de alterações comportamentais com manifestação de crises motoras límbicas que surgem aos 15-30 minutos após injeção, e podem perdurar por horas (status epilepticus), caracterizando o período agudo do modelo (Turski et al., 1983; Cavalheiro, 1995). O animal que sobrevive ao quadro agudo passa por um período latente, livre de crises comportamentais, que tem duração média de 14 dias, que termina no momento em que o animal apresenta a primeira crise espontânea que perdura por toda a vida (Leite et al., 1990).
Estudos experimentais in vitro mostraram que o efeito epileptogênico desse e de outros agentes colinérgicos resulta de descargas originadas em neurônios do hipocampo devido ao bloqueio das correntes de potássio transmembranar IM. Esse mecanismo parece explicar a maciça ativação dos neurônios do hipocampo durante o status epilepticus induzido pela pilocarpina, status esse que conduz à morte celular e reorganização sináptica do circuito hipocampal, provocando mudanças epileptogênicas permanentes. A lesão neuronal não é, portanto, decorrente de efeito tóxico da pilocarpina, mas sim do efeito excitotóxico envolvendo receptores de glutamato e influxo de ions cálcio. As crises que ocorrem durante o status epilepticus na fase aguda diferem farmacologicamente das crises espontâneas posteriores ao status, estando na base desse fato a referida reorganização dos circuitos neuronais após a ocorrência do status. As crises espontâneas que ocorrem após o status epilepticus são bastante similares, na sua farmacologia, às crises límbicas (focais/parciais) originadas pelo kindling (modelo de indução de crises e epilepsia por estimulação elétrica de áreas no sistema límbico, amígdala e hipocampo). Assim, em ambos os modelos, a carbamazepina, o fenobarbital, a fenitoína e o ácido valprico são efetivos, enquanto a etossuximida é ineficaz.
Os achados histológicos no modelo da pilocarpina revelam a presença de lesão neuronal em inúmeras áreas cerebrais, atingindo especialmente a formação hipocampal (regiões de CA1, CA3 e hilo do giro denteado), o córtex entorrinal e piriforme, e a amígdala (Turski et al., 1983; Leite et al., 1990; Cavalheiro, 2005) (Figura 1).
A epileptogênese é também acompanhada por alterações neuroquímicas e celulares que levam ao desequilíbrio entre a neurotransmissão inibitória e excitatória no hipocampo, e consequentemente à ocorrência de crises espontâneas. As alterações histológicas e moleculares são muito similares às observadas em pacientes com ELTM, com a vantagem de que podemos obtê-las em semanas enquanto no paciente demoram anos (Figura 2).
A neurogênese, ou seja, a formação de novos neurônios exerce um papel na reparação da lesão cerebral, mas essa função é comprometida pelas crises na ELTM. Estudos em modelos animais revelam que momentos após um evento precipitante inicial (crises agudas ou status epilepticus) ocorrem aumentos da neurogênese hipocampal e recrutamento anormal de neurônios recém-gerados no hipocampo. No entanto, as crises interferem com a migração, a proliferação e o desenvolvimento neuronal normal de células recém-geradas no hipocampo possivelmente formando um circuito de amplificação de crises. Estudos em modelos animais revelam que a fase crônica da epilepsia está associada com diminuição substancial da neurogênese, demonstrada também em hipocampo de pacientes com ELTM. Análises sugerem que a diminuição da neurogênese e a diminuição dramática na diferenciação neuronal de células recém-geradas são consequências do efeito prejudicial de crises espontâneas recorrentes, estando relacionadas com ativação de cascatas inflamatórias. Portanto, as estratégias terapêuticas devem ser direcionadas em minimizar a ocorrência de crises espontâneas recorrentes e melhorar as respostas neuroprotetoras (Parent et al., 2006).
Como tratar a epilepsia?
As epilepsias podem ser tratadas com drogas antiepilépticas e, em casos de refratariedade das crises ao tratamento medicamentoso, a cirurgia para remoção do foco epiléptico é uma alternativa. Existem mais de vinte drogas disponíveis no mercado atualmente e a escolha depende de fatores como o tipo de crise, a frequência com que ocorre, a idade e estilo de vida do paciente. As drogas antiepilépticas geralmente são eficazes quando administradas em monoterapia, mas admitem-se politerapias (mais de uma droga concomitantemente) quando a monoterapia falha em controlar a crise do paciente.
Em alguns casos o tratamento causa efeitos indesejáveis, sendo comum cansaço, tonturas e ganho de peso, mas quadros mais graves de reação alérgica e depressão e psicose podem ocorrer. É fundamental o acompanhamento médico durante o tratamento. A descontinuidade do tratamento pode ser indicada pelo clínico quando o paciente fica de dois a quatro anos livre de crises. A retirada abrupta do tratamento pode causar status epilepticus considerado uma emergência clínica.
O tratamento cirúrgico funciona?
Quando as crises não são controladas pelo tratamento medicamentoso aplicado em monoterapia ou politerapia, o paciente pode ser submetido a uma avaliação para a cirurgia. A avaliação é feita por equipe multiprofissional que cuidadosamente interpreta dados de exames de vídeo-EEG (registro eletrencefalográfico monitorado por vídeo), exames clínicos, de imagem (ressonância magnética e/ou tomografia computadorizada), testes neuropsicológicos para definir se o paciente é ou não candidato a cirurgia. O teste de Wada consiste no anestesiamento do hemisfério que contém a área afetada pela aplicação do amobarbital sódico na carótida interna, para analisar o hemisfério contralateral por meio de testes da fala e de memória. Esse também é usado como parâmetro na avaliação de pacientes candidatos a cirurgia. Caso o paciente apresente falha na manutenção da memória a cirurgia é contraindicada.
As epilepsias que mais se beneficiam com o tratamento cirúrgico são as do lobo temporal e a encefalite de Rasmussen. A síndrome de Rasmussen é descrita como uma encefalite progressiva causando hemiparesia, retardo mental e epilepsia de difícil controle. As taxas de cessação das crises em pacientes com epilepsia do lobo temporal que são lobectomizados variam de 70% a 90%. Outros procedimentos cirúrgicos podem ser indicados, como a calosotomia (secção do corpo caloso, feixe de fibras que liga um hemisfério a outro), indicada para tratar epilepsias severas cujas crises iniciam em um hemisfério e se propagam para o outro. Impede a generalização da crise, mas não bloqueia a crise focal. A transecção subpial múltipla é um procedimento que pode ser aplicado a casos em que a área geradora da crise não pode ser removida. O neurocirurgião faz uma série de cortes para prevenir o espraiamento das crises para outras regiões, mas as habilidades ficam preservadas (Rassi Neto et al., 2001).
É possível prevenir a epilepsia?
Muitos casos de epilepsias podem ser prevenidos com medidas simples, como usar cinto de segurança, usar cadeiras de carro para portar crianças pequenas, usar capacete quando for pedalar uma bicicleta; enfim, fazer tudo que possa prevenir traumas na cabeça. Iniciar tratamento medicamentoso adequado logo após a primeira ou segunda crise e após crises febris ajuda a prevenir epilepsia. Fazer acompanhamento pré-natal com controle de pressão alta, tratar quadros de infecção durante a gestação são medidas que podem impedir a ocorrência de lesão no cérebro do bebê e se tornar um foco epiléptico na vida adulta. Tratar doenças cardiovasculares, evitar infestação por cisticercos são medidas que ajudam a evitar epilepsia. Estudos mostram que o curso natural da epileptogênese pode ser influenciado por fatores como predisposição genética, mecanismos epigenéticos (modulado pelo meio ambiente) e uso de drogas antiepilépticas.
Existem alvos terapêuticos para a epilepsia?
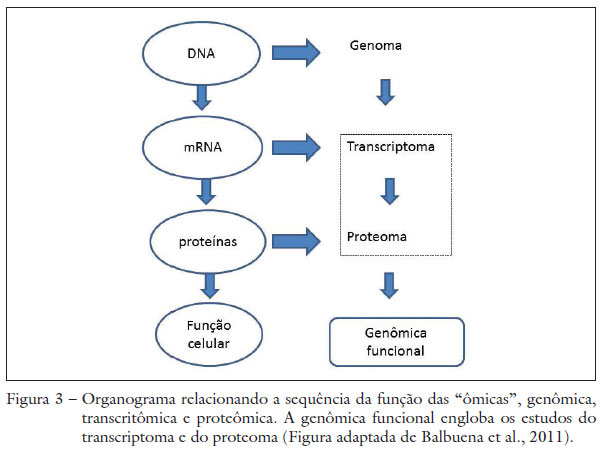
A transcriptômica e a proteômica são métodos usados para estudar o transcriptoma e o proteoma dentro do contexto da genômica funcional (Figura 3). A genômica funcional surge com o objetivo de identificar a sequência e a função de genes e de proteínas comparando-se condições patológicas com condições controles. A proteômica tem se destacado como uma metodologia essencial para investigar o papel funcional de proteínas no metabolismo celular. O termo proteômica se refere ao total de proteínas expressas por um genoma sob certa condição fisiológica (Wilkins et al., 1996). A metodologia é baseada nos princípios da bioquímica, biofísica e informática. A análise compreende quatro fases: extração, purificação, separação e identificação de proteínas diferencialmente expressas em condição normal ou patológica. Isso permite identificar as proteínas que são diferentes do tecido normal.
Resumidamente, é feita uma extração de proteínas de amostras teciduais, a amostra é aplicada a um gel de poliacrilamida para corrida eletroforética bidimensional. Primeiramente é feita a focalização isoelétrica (as proteínas separam por cargas) para em seguida serem submetidas a uma corrida em gel de poliacrilamida (SDS-PAGE) que separa as proteínas por peso molecular (o gel é acoplado a uma fonte elétrica para a separação proteica). Ao final das corridas se obtêm os spots proteicos que são selecionados e cortados do gel por meio de análise visual e de software criado para essa finalidade (PD Quest 7.3.1, Bio Rad). As Figuras 4 e 5 mostram, respectivamente, um esquema da separação bidimensional e um gel obtido com a corrida de amostras de hipocampo humano obtido por meio de cirurgia de paciente com ELTM e controle por necropsia. Os spots contendo as proteínas são submetidos a um processo de digestão por tripsina, e os peptídeos gerados são separados por HPLC (cromatografia líquida de alta performance) e analisados por espectrometria de massa (MS/MS) sendo posteriormente identificados em bancos de dados específicos (MASCOT).
A transcriptomica permite a análise de genes expressos em diferentes condições experimentais. Foi introduzida em meados dos anos 1990, porém pouco utilizada em estudos de epilepsias. Outro fator que pode ter contribuído para o pouco uso da transcriptômica em estudos de epilepsia é a dificuldade de interpretar os dados, pois é preciso ter conhecimento sobre análise dos arranjos (arrays), algoritmos de normalização e obter valores de referência para seleção dos genes, considerando as diferentes linhagens e espécies, estruturas cerebrais, insultos etc. Apesar das discrepâncias entre os achados, os estudos permitiram identificar alguns alvos que foram testados em modelo experimental de epilepsia com o intuito de alterar o curso da epileptogênese. A Eritropoietina (EPO), um agente neurotrófico, antiapoptótico, antioxidante e anti-inflamatório quando administrada após indução do SE pela pilocarpina, durante sete dias, causou redução na frequência e duração de crises, no dano da barreira hematoencefálica, na neurodegeneração, e na dispersão de células granulares no giro denteado, sendo benéfica para as alterações cognitivas associadas ao processo epiléptico. Efeitos similares foram descritos com a aplicação do BDNF e FGF-2 no modelo da pilocarpina. Agentes anti-inflamatórios não esteroidais (celecoxib) aplicados 24h após status epilepticus induzido por pilocarpina, mantidos durante 42 dias, não alteraram o desenvolvimento da epilepsia, porém reduziu a frequência e a duração das crises, causou neuroproteção no hipocampo, reduziram a dispersão no giro denteado e hilo. O uso de imunossupressores como o tacrolimus, uma droga que se liga à imunofilina intracelular para formar complexo que impede as células T de responderem à ativação do sistema imune, não altera latência, frequência e gravidade das crises (Pitkanen, 2010). Até a presente data, nenhum tratamento foi capaz de impedir ou alterar o curso da epileptogênese iniciada após um insulto ao sistema nervoso central.
Os dados obtidos com estudos de proteômica para avaliar a epilepsia também são escassos na literatura. Estudos empregando tecido hipocampal de paciente com epilepsia do lobo temporal revelam redução na concentração de várias proteínas com as seguintes funções: chaperonas (TCP-1α e HSP70), sinalização celular l signaling (MAPKK), sinalização transcripcional, componente sinaptossoma (α-synuclein) e citoesqueleto (tubulina, profilina e vinculina). Por outro lado, houve aumento na expressão de proteínas associadas com função antioxidante (peroxiredoxina 6), gliose e células endoteliais (apo A-I) (Yang et al., 2006). Alguns estudos em líquor de pacientes também foram feitos (Xiao et al., 2009).
Nosso grupo também tem trazido contribuição para a área da proteômica e epilepsia. Recentemente, Persike et al. (2012) mostraram que o hipocampo de paciente com epilepsia do lobo temporal possui menor número de spots do que o tecido controle, ou seja, menor expressão proteica (Comitê de Ética n.1692/05). De um total de 16 proteínas diferencialmente expressas entre os grupos de pacientes epilépticos e controle, apenas nove puderam ser identificadas. As proteínas aumentadas no tecido epiléptico foram indicativas de: quebra de barreira hematoencefálica; controle do estoque de neurotransmissores (H+ATPase); atividade compensatória ante processos neurodegenerativos (HSP 70); presença de comorbidades psiquiátricas (esquizofrenia). Encontram-se em fase de análise os dados obtidos com estudos de proteômica em hipocampo de ratos apresentando crises crônicas induzidas por pilocarpina, assim como de ratos resistentes ao efeito convulsivante da pilocarpina. Esses dados serão importantes para nos informar sobre os seguintes aspectos:
1) Existem similaridades nas alterações moleculares em pacientes com ELTM e modelo experimental;
2) Será possível identificar marcadores relacionados com a resistência a epilepsia;
3) É possível identificar comorbidades psiquiátricas em pacientes com epilepsia por meio do marcador proteico.
Todas essas questões serão respondidas ao término das análises.
Recebido em 15.2.2013 e aceito em 26.2.2013.
Agradecimentos - A autora agradece o apoio financeiro de Fapesp, CNPq, Capes, INNT/ MCT, e a Dra. Daniele S. Persike pelas pesquisas realizadas.
- BABB, T. L. et al. Distribution of pyramidal cell density and hyperexcitability in the epileptic human hippocampal formation. Epilepsia, v.25, n.6, p.721-8, 1984.
- ______. Synaptic reorganization by mossy fibers in human epileptic fascia dentata. Neuroscience, v.42, n.2, p.351-363, 1991.
- BALBUENA, T. S. et al. Challenges in proteome analyses of tropical plants. Brazilian J Plant Physiol, v.23, n.2, p.91-104, 2011.
- CAVALHEIRO, E. A. The pilocarpine model of epilepsy. Ital J Neurol Sci, v.16, n.1-2, p.33-7, 1995.
- FISHER, R. S. et al. Epileptic seizures and epilepsy: definitions proposed by the international League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia, v.46, n.4, p.470-2, 2005.
- KOTAGAL, P. Seizure symptomatology of temporal lobe epilepsy. In: LÜDERS, H. (Ed.) Epilepsy Surgery New York: Raven Press, p.143-156, 1991.
- KURUBA, R. et al. Hippocampal neurogenesis and neural stem cells in temporal lobe epilepsy. Epilepsy Behav v.14, n.1, p.65-73, 2009.
- LEITE, J. P. et al. Spontaneous recurrent seizures in rats: an experimental model of partial epilepsy. Neurosci. Biobehav. Rev, v.14, n.4, p.511-7, 1990.
- LOSCHER, W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure, v.20, n.5, p.359-68, 2011.
- MARCHETTI, R. L. et al. Transtornos mentais associados à epilepsia. J. Rev. Psiq. Clín v.32, n.3, p.170-182, 2005.
- MOREIRA, R. G. M. Epilepsia: concepção histórica, aspectos conceituais, diagnóstico e tratamento. Mental, Barbacena, ano II, n.3, p.107-22, 2004.
- PARENT, J. M. et al. Aberrant Seizure-Induced Neurogenesis in Experimental Temporal Lobe Epilepsy. Ann Neurol, v.59, p.81-91, 2006.
- PERSIKE, D. S. et al. Hippocampal Proteomic Profile in Temporal Lobe Epilepsy. J Epilepsy Clin Neurophysiol, v.18, n.2, p.53-6, 2012.
- PITKANEN, A. Therapeutic approaches to epileptogenesis - Hope on the horizon. Epilepsia, v.51, n.3, p.2-17, 2010.
- RASSI NETO, A. et al. Tratamento cirúrgico das epilepsias. Rev. Neurociências, v.9, n.3, p.118-26, 2001.
- SUTULA, T. P. Mechanisms of epilepsy progression: current theories and perspectives from neuroplasticity in adulthood and development. Epilepsy Res, v.60, p.161-71, 2004.
- TURSKI, W. A. et al. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res, v.9, n.3, p.315-35, 1983.
- WILKINS, M. R. et al. Current challenges and future applications for protein maps and post -translational vectors maps in proteome projects. Electrophoresis, v.17, p.830-8, 1996.
- WHO. Epilepsy. Disponível em: <http://www.who.int/mediacentre/factsheets/fs999/en/index.html>
- XIAO, F. et al. Proteomic analysis of cerebrospinal fluid from patients with idiopathic temporal lobe epilepsy. Brain res., v.1255, p.180-9, 2009.
- YACUBIAN, E. M. T. Tratamento da epilepsia na infância. Jornal de Pediatria, v.78, n.1, p.S19-S27, 2002.
- YANG, J. W. et al. Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids, v.30, p.477-93, 2006.
Datas de Publicação
-
Publicação nesta coleção
11 Abr 2013 -
Data do Fascículo
2013
Histórico
-
Recebido
15 Fev 2013 -
Aceito
26 Fev 2013