Abstracts
Palladium acetylacetonate dissolved in 1-n-butyl-3-methylimidazolium tetrafluoroborate ionic liquid catalyses the selective two-phase hydrogenation of conjugated and non-conjugated (functionalized and non-functionalized) dienes into the respective monoenes. The system does not require the use of organic solvents, the products are removed by simple decantation or distillation and the recovered ionic catalytic solution can be reused several times without any significant changes in its catalytic activity and selectivity.
biphasic hydrogenation; ionic liquids; dienes; palladium catalysts
Bis-acetilacetonato de paládio dissolvido no líquido iônico tetrafluoroborato de 1-n-butil-3-metilimidazólio catalisa de forma seletiva a hidrogenação de dienos conjugados e não-conjugados (funcionalizados ou não) às suas respectivas mono-olefinas. O sistema não requer o uso de co-solventes orgânicos, sendo os produtos da reação separados por simples decantação ou destilação. A fase iônica pode ser reutilizada até 15 vezes sem perdas significativas na sua atividade catalítica e seletividade.
Article
Pd(II)-Dissolved in Ionic Liquids: A Recyclable Catalytic System for the Selective Biphasic Hydrogenation of Dienes to Monoenes
Jairton Dupont* * e-mail: dupont@if.ufrgs.br , Paulo A. Z. Suarez, Alexandre P. Umpierre and Roberto F. de Souza
Instituto de Química, Universidade Federal do Rio Grande do Sul, Av. Bento Gonçalves, 9500 91501-970 Porto Alegre-RS, Brazil
Bis-acetilacetonato de paládio dissolvido no líquido iônico tetrafluoroborato de 1-n-butil-3-metilimidazólio catalisa de forma seletiva a hidrogenação de dienos conjugados e não-conjugados (funcionalizados ou não) às suas respectivas mono-olefinas. O sistema não requer o uso de co-solventes orgânicos, sendo os produtos da reação separados por simples decantação ou destilação. A fase iônica pode ser reutilizada até 15 vezes sem perdas significativas na sua atividade catalítica e seletividade.
Palladium acetylacetonate dissolved in 1-n-butyl-3-methylimidazolium tetrafluoroborate ionic liquid catalyses the selective two-phase hydrogenation of conjugated and non-conjugated (functionalized and non-functionalized) dienes into the respective monoenes. The system does not require the use of organic solvents, the products are removed by simple decantation or distillation and the recovered ionic catalytic solution can be reused several times without any significant changes in its catalytic activity and selectivity.
Keywords: biphasic hydrogenation, ionic liquids, dienes, palladium catalysts
Introduction
In order to minimize the amount of waste and by-products from chemical processes, the ideal chemical reaction should not only be selective but should also be just a simple addition ("atom economy") in which any other reactant is employed in catalytic amounts1. The possibility of modulating the catalyst's properties (both electronic and steric) by varying the metal centre and/or ligands places transition-metal catalyst precursors as attractive candidates for promoting such reactions1,2. However, the difficulties involving product separation from the reaction mixture and catalyst recycling, as well as the necessity of using organic solvents, are the major drawbacks of these one-phase homogeneous catalytic reactions3. It is now well accepted that liquid-liquid two-phase catalytic reactions are important alternatives to homogeneous catalyzed one-phase reactions3,4. In these systems, after the reaction, one phase contains the product and the other phase the transition-metal catalyst. The recovered catalyst phase can be reused without any further treatment. Moreover, in these two-phase systems it is possible to extract the primary products during the reaction and thus modulate the product selectivity5. Amongst the "mobile" phases employed for the immobilization of transition-metal catalyst precursors3,4,6,7 molten salts are attracting increasing and continuous interest amongst academic and industrial chemists8. One asset of biphasic catalysis in ionic liquids is that it allows in various cases the direct transposition of known homogeneous one-phase reactions to two-phase systems without the synthesis of specially designed ligands and/or complexes. Moreover, in some cases simple transition-metal salts can be used without any special ligand to stabilize the catalytically active species8f,h, l, m.
We wish to report here that palladium acetylacetonate (Pd(acac)2) immobilized in 1-n-butyl-3-methylimida-zolium tetrafluoroborate (BMI.BF4) ionic liquid is able to catalyze the selective reduction of simple and functionalized dienes to monoenes in a typical biphasic catalytic reaction. It is interesting to note that despite the importance of this transformation and ever-growing environmental concerns, only a few homogeneous or biphasic catalytic process for the selective hydrogenation of dienes into monoenes have been described9,10.
Results and Discussion
Initial investigations were performed on the hydrogenation of 1,3-butadiene (80 mmol) by Pd(acac)2 (0.08 mmol) dissolved in 3 mL of 1-n-butyl-3-methylimidazoilum tetrafluoroborate (BMI.BF4, liquid down to -81°C)8fand in 3 mL of 1-n-butyl-3-methylimidazoilum hexafluorophosphate (BMI.PF6, liquid down to -61°C)8fionic liquids at 50°C (Scheme 1 and Table 1).

The organic phase is separated from the ionic catalytic solution by simple decantation and analyzed by gas chromatography. For comparative purposes the results obtained from the hydrogenation of 1,3-butadiene by Pd(acac)2 dissolved in dichloromethane (homogeneous conditions), in liquid 1,3-butadiene (without solvent), and by Pd/C (heterogeneous conditions) have also been included in Table 1.
It is clear from Table 1, that for analogous 1,3-butadiene conversions, the selectivities in butenes achieved by Pd(acac)2 dissolved in ionic liquids are similar to those observed under homogeneous conditions (CH2Cl2) and are higher than in heterogeneous conditions or in liquid 1,3-butadiene. It is important to note that, using BMI.BF4 as solvent, the formation of butane only occurs after more than 95% of the 1,3-butadiene was converted into butenes (see Figure 1). Moreover, we observed that the formed 1-butene is isomerised in 2-butenes even in the early stages of the process, suggesting the involvement of Pd-H species similar to those formed in the one-phase hydrogenation11.
The effects of temperature, hydrogen pressure and catalyst precursor concentration in 1,3-butadiene reduction are summarized in Table 2. From these data it is clear that there are no significant changes in the selectivity by varying these reaction parameters. Moreover, the turnover frequencies (TOF) increase with increasing reaction the initial hydrogen pressure and temperature (see entries 2 and 9-11). It is also evident from Table 2 that with an increase of the catalyst precursor concentration there is a significant drop on the reaction time to achieve similar 1,3-butadiene conversions (entries 2 and 6-8). These results are similar to those observed under one-phase conditions 9a.
All these results, associated with the selectivity, suggest that the mechanism of the hydrogenation reaction for the palladium complex dissolved in the ionic liquid is analogous to the one generally accepted for the one-phase reactions (see later).
This method has been extended to a series of simple and functionalized dienes and similar selectivities in monoenes were observed (Table 3). The catalytic system can promote the selective reduction of functionalized dienes to monoenes without competitive reduction of acid, ester or nitro groups. Moreover, both conjugated and non-conjugated dienes are selectively hydrogenated to the corresponding monoenes (see for example entries 5 to 8, Table 3). Note that the hydrogenation of methyl sorbate and 1-nitro-1,3-butadiene gives the trans mono-ene almost exclusively as judged by the 1H NMR of the crude reaction product. Therefore, this reduction is highly chemo- and stereoselective and it should proceed by 1,2-addition of hydrogen to the double bond containing the more electron-donating substituent and/or the least substituted of the two double bonds. It is interesting to note that the hydrogenation of sorbic ester by palladium catalyst precursors in one-phase conditions gave usually a mixture of mono-ene (70%) and fully hydrogenated substrate(30%)9b. Moreover, it was recently reported that the biphasic hydrogenation of sorbic acid by water soluble Ru(II) complexes produces a mixture of cis and trans hexanoic acids with very low TON (up to 62)10a.
It is well known that the selectivity for hydrogenation of dienes in the presence of monoenes usually arises from the exceptional stability of the h3-allyl intermediate complexes, formed by M-H addition to the conjugated diene. In a homogenous hydrogenation mixture there is a competition between the diene and the monoene for the catalytic active site (see Scheme 2)11. The reaction pathway involving the h3-allyl intermediate is thermodynamically favoured. Consequently, the diene in the reaction media is almost completely hydrogenated before the concentration of the monoene increases to the point that it gains access to the metal center. In the case of biphasic catalysis in ionic liquids a similar pathway can explain the observed results. Moreover, in these cases, the high selectivity in monoenes can also be attributed to the higher concentration of the diene in the ionic phase, where the reaction is presumed to take place, as compared to the monoene. Indeed, 1,3-butadiene is almost four times more soluble in the ionic phase than butenes (4.4 wt % (1,3-butadiene) and 1.0 wt % (butenes) at 25°C and 771 mmHg) in BMI.BF4 and in BMI.PF6 the solubility of 1,3-butadiene and butenes are 2.2 wt % and 1.0 wt % at 25°C and 771 mmHg, respectively.

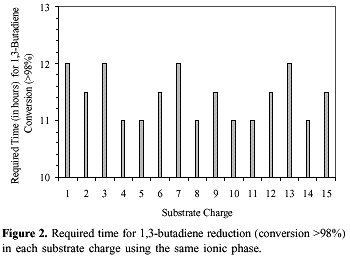
Recycling of the ionic catalytic phase was achieved by simple phase separation and the recovered yellow ionic viscous solution was reused for further runs. In the case of 1,3-butadiene this ionic solution could be reused for more than 15 runs with complete conservation of the catalytic activity and selectivity (see Figure 2). The palladium content in the organic phase was less than 0.01% as determined by atomic absorption analysis. However, in the case of sorbic acid (entry 3, Table 2) the formation of metallic palladium was observed and therefore rendered catalyst reuse impractical. The catalyst decomposition, in this case, is probably due to the extraction of palladium species from the ionic solution by the organic acid and then reduced to metallic palladium. Indeed, the catalyst extraction and decomposition is completely suppressed when the methyl ester is used as substrate (entries 1 and 2, Table 2).
The method does not normally use an organic solvent but, if necessary, is achievable using diisopropyl ether as cosolvent with viscous methyl sorbate, 1-nitro-1,3-butadiene and sorbic acid (Table 3, entries 2, 3 and 4).
In summary this new "biphasic" process (solvent-free and recyclable) is not only economically viable and applicable to large-scale reactions, but is also environmentally friendly.
Experimental
General
All manipulations were performed under dry, oxygen-free argon using standard Schlenk tube techniques. All solvents were dried over suitable dessicant agents and distilled under argon prior to use. Infrared (nujol mulls or KBr pellets) spectra were recorded in the 4000 - 400 cm-1 region using a Mattson 3020 FTIR spectrophotometer. 1H and 13C {1H} NMR spectra were recorded at 200.13 and 50.32 MHz, respectively, using a Varian VXR-200 instrument. Proton and carbon shifts (d/ppm, J/Hz) are positive down field relative to external SiMe4. Elemental analyses were carried out by the "Central Analítica IQ/UFRGS" (Porto Alegre, Brazil). Mass spectra were obtained with a GC-MS HP5988A (EI, 70 eV). The metal contents in both ionic and organic phases were determined by atomic absorption analysis using a Perkin Elmer 2380, HGA 400, atomic absorption spectrometer with graphite tube atomizer. The substrate conversions and product selectivities were analyzed by gas chromatography on a Varian 3400 CX chromatograph equipped with an LM100 column 20 m x 0.25 mm x 0.2 mm, equipped with a FID detector; N2 was the carrier (1 mL-1); the temperature program was from 35°C (5 min) to 200°C (15 min) at a heating rate of 10°C min-1, using cyclohexane as internal standard. The catalytic reactions were performed in a 100 mL stainless steel autoclave, containing a magnetic bar, temperature indicator and a manometer. The consumption of hydrogen was monitored by the decrease of the reactor's pressure.
Materials
The ionic liquids were prepared according to literature procedures8a. 1,3-Butadiene was distilled under argon and stored over molecular sieves (3 Å). 1-Nitro-1,3-butadiene was prepared according to procedures described in the literature12. All other reagents were obtained from commercial sources and were used as received without further purification.
Typical Procedure for the Hydrogenation Reaction
The ionic liquid (3 mL) and the transition metal catalyst precursor were mixed in a Schlenk tube and then transferred into the autoclave under argon. The substrate (10 mL) was introduced and the reactor was pressurized with hydrogen at the desired temperature. The consumption of hydrogen was monitored by the decrease of the reactor pressure. For the reaction, a well-stirred system is required, since the mass transfer between the phases is an important parameter in these systems (in the absence of stirring the consumption of hydrogen is completely stopped). The reaction was stopped after the desired time and the organic phase analyzed by gas chromatography. After the reaction, the manipulations and separations of the phases were performed under ambient conditions and, when 1,3-butadiene was used, the temperature was lowered to _20°C. In cases where the ionic phase was reused, this procedure was carried out under argon. The reactions were repeated at least twice in order to ensure reproducibility of the system. For 1-nitro-1,3-butadiene and methyl sorbate reactions, the products were extracted by washing the reaction mixtures with diisopropyl ether (5 x 15 mL) and analyzed CG-MS and 1H NMR.
Acknowledgements
We thank CNPq and FAPERGS for support of this work. We are grateful to Dr. John Spencer (TRI, England) for proofreading the manuscript.
References
1. Trost, B. M. Angew. Chem. Int. Ed. 1995, 34, 259.
2. Herrmann, W. A.; Cornils, B. Angew. Chem. Int. Ed. 1997, 36, 1049.
3. Aqueous-Phase Organometallic Catalysis, Concepts and Applications; Cornils, B.; Herrmann, W. A. (Eds), Wiley-VCH: Weinheim, 1996.
4. Driessen-Hölscher, B. Adv. Catal., 1998, 42, 473.
5. Prinz, T.; Keim, W.; Driessen-Hölscher, B. Angew. Chem. Int. Ed. 1996, 35, 1708.
6. (a) Horváth, I. T.; Kiss, G.; Cook, R. A.; Bond, J. E.; Stevens, P. A.; Rabai, J.; Mozeleski, E. J. J. Am. Chem. Soc. 1998, 120, 3133. (b) Klement, I.; Lütjens, H.; Knochel, P. Angew. Chem. Int. Ed. Engl. 1997, 36, 1454. (c) Fish, R. H. Chem. Eur. J. 1999, 5, 1677.
7. (a) Kainz, S.; Koch, D.; Baumann, W.; Leitner, W.; Angew. Chem. Int. Ed. 1997, 36, 1628. (b) Jessop, P. G.; Ikariya, T.; Noyori, R. Chem. Rev. 1999, 99, 475.
8. (a) Suarez, P. A. Z.; Dullius, J. E. L.; Einloft, S.; de Souza, R. F.; Dupont, J. Polyhedron 1996, 15, 1217. (b) Chauvin, Y.; Mussmann, L.; Olivier, H. Angew. Chem. Int. Ed. 1995, 34, 2698. (c) Suarez, P. A. Z.; Dullius, J. E. L.; Einloft, S.; de Souza, R. F.; Dupont, J. Inorg. Chem. Acta. 1997, 255, 207. (d) Müller, L. A.; Dupont, J.; de Souza, R. F. Macromol. Rap. Commun. 1998, 19, 409. (e) Dyson, P. J.; Ellis, D. J.; Parker, D. G.; Welton, T. Chem. Commun. 1999, 25. (e) Monteiro, A. L.; Zinn, F. K.; de Souza, R. F.; Dupont, J. Tetrahedron: Asymmetry 1997, 8, 177. (f) Dullius, J. E. L.; Suarez, P. A. Z.; Einloft, S.; de Souza, R. F.; Dupont, J.; Fischer, J.; de Cian, A. Organometallics 1998, 17, 815. (g) Zim, D.; de Souza, R. F.; Dupont, J.; Monteiro, A. L. Tetrahedron Lett. 1998, 39, 7071. (h) Kaufmann, D. E.; Nouroozian, M.; Henze, H. Synlett 1996, 1091. (i) Herrmann, W. A.; Böhm, V. P. W. J. Organomet. Chem. 1999, 572, 141. (j) Welton, T. Chem. Rev., 1999, 99, 2071. (l) Carmichael, A. J.; Earle, M.; Holbrey, J. D.; McCormac, P. B.; Seddon., K. R. Org. Lett. 1999, 1, 997. (m) de Bellefon, C.; Pollet, E.; Grenouillet, P. J. Mol. Catal. A: Chem 1999, 145, 121.
9. (a) Parshall, G. W.; Ittel, S. D. Homogeneous Catalysis - The Applications and Chemistry of Catalysis by Soluble Transition Metal Complexes; John Wiley & Sons, Inc.; New York, 1992, p 31-42. (b) Cho, I. S.; Alper, H. Tetrahedron Lett. 1995, 36, 5673. (c) Kameda, N.; Igarashi, R. Bull. Chem. Soc. Jpn. 1994, 6, 577. (d) Vasilev, A. A.; Serebryakov, E. P. Mendeleev Commun. 1994, 4. (e) Tsuji, J.; Sozuki, H. Chem. Lett. 1977, 1083. (f) Fahey, D. R. J. Org. Chem. 1973, 38, 3343.
10. (a) Driessen-Hölscher, B.; Heinen, J. J. Organomet. Chem. 1998, 570, 141. (b) Halpern, J.; Pribanic, M. Inorg. Chem. 1970, 9, 2616.
11. Parshall, G. W. J. Am. Chem. Soc. 1972, 94, 8716.
12. Bloom, A. J.; Mellor, J. M. Tetrahedron Lett. 1986, 27, 873.
Received: December 6, 1999
- 1. Trost, B. M. Angew. Chem. Int. Ed. 1995, 34, 259.
- 2. Herrmann, W. A.; Cornils, B. Angew. Chem. Int. Ed. 1997, 36, 1049.
- 3. Aqueous-Phase Organometallic Catalysis, Concepts and Applications; Cornils, B.; Herrmann, W. A. (Eds), Wiley-VCH: Weinheim, 1996.
- 4. Driessen-Hölscher, B. Adv. Catal., 1998, 42, 473.
- 5. Prinz, T.; Keim, W.; Driessen-Hölscher, B. Angew. Chem. Int. Ed. 1996, 35, 1708.
- 6. (a) Horváth, I. T.; Kiss, G.; Cook, R. A.; Bond, J. E.; Stevens, P. A.; Rabai, J.; Mozeleski, E. J. J. Am. Chem. Soc 1998, 120, 3133.
- (b) Klement, I.; Lütjens, H.; Knochel, P. Angew. Chem. Int. Ed. Engl 1997, 36, 1454.
- (c) Fish, R. H. Chem. Eur. J. 1999, 5, 1677.
- 7. (a) Kainz, S.; Koch, D.; Baumann, W.; Leitner, W.; Angew. Chem. Int. Ed. 1997, 36, 1628.
- 8. (a) Suarez, P. A. Z.; Dullius, J. E. L.; Einloft, S.; de Souza, R. F.; Dupont, J. Polyhedron 1996, 15, 1217.
- (b) Chauvin, Y.; Mussmann, L.; Olivier, H. Angew. Chem. Int. Ed. 1995, 34, 2698. (c) Suarez, P. A. Z.; Dullius, J. E. L.; Einloft, S.; de Souza, R. F.; Dupont, J. Inorg. Chem. Acta. 1997, 255, 207.
- (d) Müller, L. A.; Dupont, J.; de Souza, R. F. Macromol. Rap. Commun. 1998, 19, 409. (e) Dyson, P. J.; Ellis, D. J.; Parker, D. G.; Welton, T. Chem. Commun. 1999, 25.
- (e) Monteiro, A. L.; Zinn, F. K.; de Souza, R. F.; Dupont, J. Tetrahedron: Asymmetry 1997, 8, 177. (f) Dullius, J. E. L.; Suarez, P. A. Z.; Einloft, S.; de Souza, R. F.; Dupont, J.; Fischer, J.; de Cian, A. Organometallics 1998, 17, 815.
- (g) Zim, D.; de Souza, R. F.; Dupont, J.; Monteiro, A. L. Tetrahedron Lett. 1998, 39, 7071. (h) Kaufmann, D. E.; Nouroozian, M.; Henze, H. Synlett 1996, 1091.
- (i) Herrmann, W. A.; Böhm, V. P. W. J. Organomet. Chem. 1999, 572, 141. (j) Welton, T. Chem. Rev., 1999, 99, 2071.
- (l) Carmichael, A. J.; Earle, M.; Holbrey, J. D.; McCormac, P. B.; Seddon., K. R. Org. Lett 1999, 1, 997.
- 9. (a) Parshall, G. W.; Ittel, S. D. Homogeneous Catalysis - The Applications and Chemistry of Catalysis by Soluble Transition Metal Complexes; John Wiley & Sons, Inc.; New York, 1992, p 31-42.
- (b) Cho, I. S.; Alper, H. Tetrahedron Lett 1995, 36, 5673. (c) Kameda, N.; Igarashi, R. Bull. Chem. Soc. Jpn. 1994, 6, 577.
- (d) Vasilev, A. A.; Serebryakov, E. P. Mendeleev Commun. 1994, 4. (e) Tsuji, J.; Sozuki, H. Chem. Lett. 1977, 1083.
- 10. (a) Driessen-Hölscher, B.; Heinen, J. J. Organomet. Chem. 1998, 570, 141.
- 11. Parshall, G. W. J. Am. Chem. Soc 1972, 94, 8716.
- 12. Bloom, A. J.; Mellor, J. M. Tetrahedron Lett. 1986, 27, 873.
Publication Dates
-
Publication in this collection
06 Nov 2000 -
Date of issue
June 2000
History
-
Received
06 Dec 1999