Abstracts
Slurry sampling in combination with flame atomic absorption spectrometry was employed for the direct determination of four essential trace elements, namely Cu, Fe, Mn and Zn in Antarctic krill. The effect of instrumental operating conditions and slurry sampling preparation on the analytical signal was investigated. For the determination of Cu, Fe and Zn, samples were suspended in a solution containing 2 mol L-1 HNO3. In the case of Mn, 4 mol L-1 HNO3 was necessary for the preparation of the slurry. The precision between sample replicates was better than 5%. The method was applied to the direct determination of Cu, Fe, Mn and Zn in Antarctic krill samples using aqueous reference solutions to prepare the calibration curves. The results obtained were in good agreement with those achieved by FAAS and ICP-AES after microwave-assisted wet digestion of the krill samples. The detection limits were 4.5, 1.0, 4.9 and 8.4 mug L-1 for Cu, Zn, Mn and Fe, respectively.
Antarctic krill; slurry; FAAS; ICP-AES
A técnica de amostragem de suspensão aliada à espectrometria de absorção atômica com chama foi empregada para a determinação direta de Cu, Fe, Mn e Zn em matrizes de Krill antártico. O efeito dos parâmetros instrumentais e o preparo das suspensões foram estudados em função do sinal analítico. Para a determinação de Cu, Fe e Zn, as amostras foram suspensas em uma solução de HNO3 2,0 mol L-1 e para a determinação de Mn, o krill foi suspenso em uma solução de HNO3 4,0 mol L-1. A precisão entre as replicas foi melhor que 5 %. A metodologia foi aplicada para a determi-nação direta de Cu, Fe, Mn e Zn em amostras de krill antártico, usando padrões aquosos no preparo das curvas analíticas. Os resultados obtidos estão concordantes com os encontrados na determinação por FAAS e ICP-AES, depois da digestão das amostras em um forno de microondas. Os limites de detecção foram 4,5; 1,0; 4,9 e 8,4 mig L-1 para Cu, Zn, Mn e Fe, respectivamente.
Article
Direct Analysis of Antarctic Krill by Slurry Sampling: Determination of Copper, Iron, Manganese and Zinc by Flame Atomic Absorption Spectrometry
Flávia L. Alvesa, Patricia Smichowskib, Silvia Faríasb, Julieta Marreroc and Marco A. Z. Arrudaa* e-mail: zezzi@iqm.unicamp.br
aDepartamento de Química Analítica, Universidade Estadual de Campinas-UNICAMP, CP 6154, 13.083-970, Campinas - SP, Brazil
bComisión Nacional de Energía Atómica, Unidad de Actividad Química, Centro Atómico Constituyentes, Av. Libertador 8250, 1429-Buenos Aires, Argentina
cComisión Nacional de Energía Atómica, Unidad de Actividad Geología, Centro Atómico Ezeiza, Av. Libertador 8250, 1429-Buenos Aires, Argentina
A técnica de amostragem de suspensão aliada à espectrometria de absorção atômica com chama foi empregada para a determinação direta de Cu, Fe, Mn e Zn em matrizes de Krill antártico. O efeito dos parâmetros instrumentais e o preparo das suspensões foram estudados em função do sinal analítico. Para a determinação de Cu, Fe e Zn, as amostras foram suspensas em uma solução de HNO3 2,0 mol L-1 e para a determinação de Mn, o krill foi suspenso em uma solução de HNO3 4,0 mol L-1. A precisão entre as replicas foi melhor que 5 %. A metodologia foi aplicada para a determi-nação direta de Cu, Fe, Mn e Zn em amostras de krill antártico, usando padrões aquosos no preparo das curvas analíticas. Os resultados obtidos estão concordantes com os encontrados na determinação por FAAS e ICP-AES, depois da digestão das amostras em um forno de microondas. Os limites de detecção foram 4,5; 1,0; 4,9 e 8,4 mg L-1 para Cu, Zn, Mn e Fe, respectivamente.
Slurry sampling in combination with flame atomic absorption spectrometry was employed for the direct determination of four essential trace elements, namely Cu, Fe, Mn and Zn in Antarctic krill. The effect of instrumental operating conditions and slurry sampling preparation on the analytical signal was investigated. For the determination of Cu, Fe and Zn, samples were suspended in a solution containing 2 mol L-1 HNO3. In the case of Mn, 4 mol L-1 HNO3 was necessary for the preparation of the slurry. The precision between sample replicates was better than 5%. The method was applied to the direct determination of Cu, Fe, Mn and Zn in Antarctic krill samples using aqueous reference solutions to prepare the calibration curves. The results obtained were in good agreement with those achieved by FAAS and ICP-AES after microwave-assisted wet digestion of the krill samples. The detection limits were 4.5, 1.0, 4.9 and 8.4 mg L-1 for Cu, Zn, Mn and Fe, respectively.
Keywords: Antarctic krill, slurry, FAAS, ICP-AES
Introduction
Most analyses by flame and plasma atomization techniques are carried out on solutions using continuous nebulization to introduce the sample. In this context, the mineralization of solid samples which is necessary prior to the analysis, which increases both the time of analysis and the risk of sample contamination inherent in the dissolution of samples. For these reasons, there is considerable interest in the development of analytical methods which allow the direct analysis of solid samples. In recent years, numerous methods have been developed in order to introduce solid samples into a variety of sources for subsequent elemental analysis1,2.Electrothermal atomization, laser ablation and glow discharge have been employed to quantify minor and trace elements in solid materials. However, a number of factors must be considered to obtain an analytical performance comparable to that achieved with liquid sampling. The introduction of the sample as a slurry is an alternative that has been used to analyze a diversity of elements in different matrices3-8. Comprehensive reviews on this methodology have been reported in the literature9,10. Slurry sampling is now an accepted technique and a very convenient way to analyze solid samples.
The slurry sample technique arose in 1974, developed by Brady et al.11,12 and combines the advantages of solid and liquid sampling13. The main advantages of the slurry sampling technique, through direct solid sample atomization, include simplification in the prior sample treatment, time reduction in sample decomposition, minimization of contamination and analyte losses, reduction of costs and reagent consumption, as well as avoiding the use of hazardous chemicals.
Particle size and the lack of homogeneity in the sample can especially affect the precision and accuracy of the analysis, accomplished through the slurry technique. Particle size is a critical factor in slurry preparation and must be optimized to a particle diameter that will also contribute to good sample agitation14. The limited range of slurry concentration is a drawback that must also be considered. This technique permits the use of aqueous standards and/or methods of standard addition10,15 to be applied for quantification.
The slurry technique associated with flame atomic absorption spectrometry (FAAS) was first applied by Willis,16 who determined several metals in geological samples. Some problems can occur in the flame; the principal difficulty is related to the slurry particle transport through the nebulizer.16-21 In order to solve these problems related to the transport efficiency of the suspended particles in the slurry, some authors proposed the use of special nebulizers.22-25
Krill (Euphasia superba) is a small shrimp extremely abundant in the Southern Ocean that can vary in length from less than 1 cm to ca. 15 cm, being mostly transparent, with large black eyes, a brught red shell, and often a vividly green stomach, but it becomes curled up and opaque when dead. The importance of krill sample analysis is based on the fact that this crustacean is a prime food source for seals, penguins and whales and these higher animals probably would not survive in the absence of the krill. The krill can accumulate trace elements and other chemicals26 and this shows promise for use as an indicator of ongoing contamination in the most pristine continent of the planet. Finally, krill is also consumed by humans, more specifically, in countries such as Chile, Russia, Japan, Poland, South Korea and Ukraine26.
The Cu, Zn, Mn and Fe determinations in krill samples are based on the importance of these elements in animal and human nutrition. Also, these elements constitute some of those that are included in the certification program for Antarctic samples. In this way, the aim of this study is to explore the development of a rapid and simple slurry method to determination of Cu, Fe, Mn and Zn in Antarctic krill and to compare the results obtained with those performed in digested krill samples by flame atomic absorption spectrometry and inductively coupled plasma atomic emission spectrometry (ICP-AES). The analysis was carried out on a certified reference material (MURST-ISS-A2). This material was prepared in the framework of the Programma Nazionale per la Richerca in Antarctica (PNRA, Italian National Program for Antarctic Research) and was coordinated by the Istituto Superiore di Sanità (ISS, Itálian National Institute of Health).
Experimental
Instrumentation
A Perkin-Elmer model AAnalyst 300 flame atomic absorption spectrometer, equipped with a deuterium lamp background correction system (Norwalk, CT, USA) was used throughout this work for the analysis of slurries and digested krill samples. Hollow cathode lamps (Perkin-Elmer, Darmstadt, Germany) of Cu, Mn, Fe and Zn were used as the radiation source and the analytical measurements were based on time averaged absorbance. The principal FAAS operating conditions are summarized in Table 1.
A Perkin-Elmer (Norwalk, CT, USA) ICP 400 sequential inductively coupled Ar plasma atomic emission spectrometer was used for the analysis of mineralized krill samples. Instrumental operating conditions are summarized in Table 2.
Solutions of Antarctic Krill were prepared by acid-assisted microwave (MW) digestion using a MLS-2000 (Milestone-FKV, Sorisole, Bergamo, Italy) equipped with temperature and pressure sensors, TFM (tetrafluoromethaxil) digestion bombs and with a magnetron of 2450 MHz.
A Cole-Parmer model 8890 ultrasonic bath (Vernon Hills, Illinois 60061, USA) was used for homogenization of the slurry prior to its introduction into the nebulizer system.
The material to be analyzed was observed under a Philips 515 (Netherlands) scanning electron microscope (SEM) that was operated at 30 kV for secondary electron (SE) imaging.
Standards and reagents
Welding Ar from AGA (Buenos Aires, Argentina) was found to be sufficiently pure for Cu, Mn, Fe and Zn ICP-AES determination. Acetylene from Air Liquid (Campinas, SP, Brasil) was employed for FAAS measurements.
All solutions were prepared with analytical quality chemicals (Merck, Darmstadt, Germany) and distilled/deionized water was used throughout this work. The HNO3 used for sample digestion and slurry preparation was distilled in a quartz sub-boiling still (Marconi, Piracicaba, Brasil). The H2O2 used for sample mineralization was of Suprapure grade (Merck, Darmstadt, Germany). For FAAS measurements a 1000 mg L-1 of Zn(II) [from Zn(NO3)2] and Fe(III) [from Fe(NO3)3] solutions were prepared as stock solutions. Cu and Mn stock solutions were prepared from their metallic forms (99.99% of purity). The standard solutions for Cu, Fe and Zn were prepared daily by making serial dilutions with 2 mol L-1 HNO3 and with 4.0 mol L-1 for Mn. For ICP-AES, the standards were prepared from Titrisol (Merck) flask.
Sample preparation procedures
Two different sample introduction approaches (slurry sampling and nebulization of solutions) were set up and compared to evaluate the possibility of using slurry sampling as a valid alternative for the determination of trace metals in Antarctic krill.
Slurry preparation of krill samples
The slurry was prepared by weighing 250 mg of lyophilized krill in a volumetric flask and completing the volume to 25 mL with 2.0 mol L-1 HNO3 for Cu, Fe and Zn. In the case of the Mn, the slurry was prepared by weighing 200 mg of lyophilised krill in a volumetric flask and completing the volume to 10 mL with 4.0 mol L-1 HNO3. The slurries were sonicated for 15 min in an ultrasonic bath (60 Hz) and Cu, Fe, Mn and Zn were quantified by FAAS. A blank was carried through out the procedure described above. Aqueous-based calibration curves were used for quantification of these trace elements.
Mineralization of krill samples
Samples of krill were also treated using an assisted acid digestion method. A 500 mg portion of sample was inserted into the microwave digestor, 6 mL HNO3 (conc.) plus 1 mL H2O2 (30 % v/v) were added and the solution was heated only after the initial reaction had subsisted. The microwave program for the decomposition of krill samples was optimized to be as short as possible and is detailed in Table 3. The average microwave power applied during the digestion cycles varied from 250 to 650 W and the time for complete sample digestion was less than 20 min.
After the decomposition, the volume was reduced to a minimum (in a ceramic plate for ca. 20 min) and then the sample was transferred to the volumetric flasks and diluted to 50 mL with 0.014 mol L-1 HNO3.
FAAS and ICP-AES measurements
FAAS: Slurried samples and solutions were measured relative to calibration standards of the individual element made up in HNO3. Absorbance time average measurements were employed to quantify the atomic signal of the four elements analyzed.
ICP-AES: Measurement conditions were optimized on signal-to-background ratio. Concentrations of Cu, Fe, Mn and Zn were calculated using the straight calibration graphs based on multielemental acidified standard solutions. In both cases, five vials were analyzed and each sample was measured three times.
Results and Discussion
Initial experiments were carried out by preparing a 1 % (m/v) suspension of krill for Cu, Zn and Fe and 2 % (m/v) for Mn in order to optimize different parameters.
Nitric acid has been used by several authors27-29 because the analytes are mobilized from the solid particles to the liquid media, which improves the precision of the method7, while nitrates do not interfere in the atomization paths for the metals involved. The concentrations selected, 2.0 mol L-1 for Cu, Fe and Zn and 4.0 mol L-1 for Mn, were used according to previous studies shown in Figure 1. The HNO3 concentration selected for Cu, Fe and Zn was based principally on the results obtained for Fe that presented a significant increase when HNO3 2.0 mol L-1 was employed in comparision to 0.014 mol L-1. As good results were already obtained for Cu and Zn, the 2.0 mol L-1 acid concentration was fixed for slurry preparation. In the case of Mn, no increase in sensibility was observe when HNO3 4.0 or 6.0 mol L-1 was used; the lower concentration was selected for security and cost reasons.
Stability of the slurry
There are several critical factors in slurry preparation that must be taken into account, such as, the stability and maximum concentration of the slurry, the particle diameter and the density of the material30. The density and the diameter of particles can be the most relevant factors, because these data are used to calculate the number of particles in suspension. When particle size decreases, there will be more particles in suspension, thus sampling errors will be reduced30. A compromise situation must be reached because if the mass of the slurry is too high, the matrix effect will affect the analytical signal. Some researchers have reported the necessity of small particle size (< 25 mm)30 for slurry analysis. The particles size of krill samples (vial N° 1070) was examinate by scanning electron microscopy (SEM), as shown in Figure 2. The material contained mostly particles of approximately 100 mm. Smaller particles (20 - 40 mm) were also observed.
Selectivity and chemical interferences
Mineralized samples of krill were analyzed by FAAS and ICP-AES and the results were compared with those obtained by slurry sampling. Although good agreement of the results was obtained for Cu, Mn, Fe and Zn with both techniques.
The results obtained for the four elements examined demonstrate that the release of a specific element from the solid phase will depend on the form in which this element is present in the matrix to be analyzed.
Krill slurry analysis
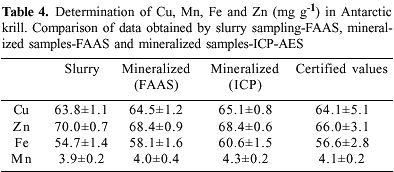
Some difficulties were observed in the determination of trace elements in krill in suspension. After mixing the lyophilized krill with 2.0 and 4.0 mol L-1 HNO3 and subsequent application of an ultrasonic treatment, a suspension containing gelatinous particles was observed. Conventional flame nebulization systems are prone to blocking when these slurries are introduced and ocassionaly the nebulizer was clogged during the preliminary studies. As a consequence, atomization efficiency was significantly reduced and losses in sensitivity occurred as the entire sample could not be atomized in the flame. To minimize these problems a cleaning step was made between each sample, using deionized water and a 2% (v/v) HNO3 solution. Although this cleaning step increased the analysis time (ca. 10%), it guaranteed better precision in the trace element determinations. Table 4 reports the final results for Cu, Fe, Mn and Zn determinations using the optimized slurry sampling method by comparing them with those obtained using FAAS and ICP-AES after microwave assisted acid digestion. In all cases the concentration values appear to agree fairly well. These results clearly demonstrate the absence of matrix effects and justify the use of aqueous standards in the calibration.
Analytical characteristics
The detection and quantification limits were calculated following the IUPAC rules31 by ten measurements of the different blank signals. The detection and quantification limits for FAAS ranged between 1.0 and 8.4 mg L-1 and 3.5 - 28.0 mg L-1, respectively. For ICP-AES the detection limits ranged from 0.5 - 4.2 mg L-1 and the quantification limits from 1.7 - 13.8 mg L-1. To evaluate the accuracy of the method the results obtained were compared with the certified values as shown in Table 4. No significant differences were observed at the 95% confidence interval using the t-test. RSDs can be considered satisfactory especially if one considers the complexity of the matrix and ranged between 0.4 - 5.0 for repeatability and 2.1 - 8.0 for reproducibility.
Conclusions
The suspension of krill samples in nitric acid solutions is a suitable method for the determination of Cu, Fe, Mn and Zn at trace levels by FAAS. In particular, the comparison of the results obtained with the certified values was most encouraging. The agreement between the analyses of digested krill samples by FAAS and ICP-AES can be considered satisfactory. The precision obtained can also be considered fairly good especially if we take into account the low concentrations of the analytes and the complexity of the matrix analyzed. A simple calibration with aqueous standards is another advantage to highlight.
Although the analysis time has been increased due to the cleaning step (the total time including sample preparation + analysis of each sample is ca. 18 min) the procedure is simple, reliable and not expensive, opening the possibility for use in certification programs. Further studies will be performed with this approach in the future for the analysis of other Antarctic matrices.
Acknowledgements
The authors are grateful to Sergio Caroli (Istituto Superiore di Sanità, ISS, Rome, Italy) for supplying the krill samples and Ana. G. Leyva (Comisión Nacional de Energía Atómica, Buenos Aires, Argentina) for the SEM micrographs. F. L. A and M. A. Z. A. are grateful to the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília, Brazil) for fellowships and to the Fundação de Amparo à Pesquisa do Estado de S. Paulo (FAPESP, São Paulo, Brazil), grant number 98/16548-3 for financial support. We also thank Prof. C. H. Collins for language assistance.
Received: September 17, 1999
FAPESP helped in meeting the publication costs of this article.
- 1. Hirata, T.; Akagi, T.; Masuda, A. Analyst 1990, 115, 1329.
- 2. Caroli, S. ICP Inform. Newslett. 1994, 20, 49.
- 3. Ebdom, L.; Wilkinson, J. R. J. Anal. At. Spectrom. 1987, 2, 325.
- 4. Miller-Ihli, N. J. Fresenius´ J. Anal. Chem. 1990, 337, 271.
- 5. Cabrera, C.; Madrid, Y.; Cámara, C. J. Anal. At. Spectrom 1994, 9, 1423.
- 6. Januzzi, G. S. B.; Krug, F. J.; Arruda, M. A. Z. J. Anal. At. Spectrom. 1997, 12, 375.
- 7. Bermejo-Barrera, P.; Moreda-Pińeiro, A.; Moreda-Pińeiro, J.; Bermejo-Barrera, A. Talanta 1998, 45, 1147.
- 8. Mierzwa, J.; Sun, Y. -Ch.; Yang, M. -H. Spectrochim. Acta Part B, 1998, 53, 63.
- 9. Bendicho, C.; De Loos-Vollebregt, M. T. C. J. Anal. At. Spectrom. 1991, 6, 353.
- 10. Miller-Ihli, N. J. Anal. Chem 1992, 64, 964 A.
- 11. Brady, D. V.; Montalvo, J. G.; Jung, J.; Curran, R. A. At. Absorpt. Newslett. 1974, 13, 118.
- 12. Brady, D. V.; Montalvo, J. G.; Glowacki, G.; Pisciotta, A. Anal. Chim. Acta 1974, 70, 448.
- 13. Stephen, S. C.; Littlejohn, D.; Ottaway, J. M. Analyst 1985, 110, 1147.
- 14. Arruda, M. A. Z.; Gallego, M.; Valcárcel, M. Quim. Anal. 1995, 14, 17.
- 15. Langmyr, F. J. Analyst 1979, 104, 993.
- 16. Willis, J. B. Anal. Chem 1975, 47, 1752.
- 17. Fuller, C. W. Analyst 1976, 101, 961.
- 18. O'Reilly, J. E.; Hickis, D. G. Anal. Chem 1979, 51, 1905.
- 19. Alder, J. F.; Backlow, P. L. At. Absorpt. Newslett 1979, 18, 123.
- 20. Stupar, J.; Ajlec, R. Analyst 1982, 107, 144.
- 21. Petrucci, G.; Van Loon, J. C. Fresenius' Z. Anal. Chem 1987, 326, 345.
- 22. Fry, R. C.; Denton, M. B. Anal. Chem 1977, 49, 1413.
- 23. Suddendorf, R. F.; Boyer, K. W. Anal. Chem 1978, 50, 1769.
- 24. Wolcott, J. F.; Sobel, C. B. Appl. Spectrosc 1978, 32, 591.
- 25. Mohamed, M.; Fry, R. C. Anal. Chem 1981, 53, 450.
- 26. Caroli, S.; Senofonte, O.; Caimi, S.; Pucci, P.; Pawels, J.; Kramer, G. N. Fresenius´J. Anal. Chem. 1998, 360, 410.
- 27. Miller-Ihli, N. J. Fresenius' J. Anal. Chem 1993, 345, 482.
- 28. Vińas, P.; Camplillo, N.; López-García, I.; Hernández-Córdoba, M. Fresenius' J. Anal. Chem. 1994, 349, 306.
- 29. Vińas, P.; Camplillo, N.; López-García, I.; Hernández-Córdoba, M. Talanta 1995, 42, 527.
- 30. Miller-Ihli, N. J. J. Anal. Atom. Spectrom 1994, 9, 1129.
-
31Analytical Methods Committee Analyst 1987, 112, 199.
e-mail:
Publication Dates
-
Publication in this collection
17 Nov 2000 -
Date of issue
Aug 2000
History
-
Received
17 Sept 1999