
Abstracts
Current studies concerning the use of acylsilanes in a variety of organic synthetic routes and the improved methodologies of their preparation have turned organosilanes into important reagents for organic chemistry. This review discusses the recent employment of acylsilanes in organic synthesis and also effective methods for their preparation.
organosilicon compounds; Brook rearrangement; carbonyl compounds; stereocontrol
Estudos sobre o emprego de acilsilanos em rotas sintéticas e as novas metodologias de preparação desenvolvidas nos últimos anos, tornaram estes organo-silanos importantes reagentes para síntese de compostos orgânicos. Esta revisão apresenta algumas aplicações recentes e vários métodos desenvolvidos para síntese de acilsilanos.
Review
Acylsilanes and Their Applications in Organic Chemistry
Amauri F. Patrocínio and Paulo J. S. Moran * * e-mail: e-mail: moran@iqm.unicamp.br
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13083-970 Campinas - SP, Brazil
Estudos sobre o emprego de acilsilanos em rotas sintéticas e as novas metodologias de preparação desenvolvidas nos últimos anos, tornaram estes organo-silanos importantes reagentes para síntese de compostos orgânicos. Esta revisão apresenta algumas aplicações recentes e vários métodos desenvolvidos para síntese de acilsilanos.
Current studies concerning the use of acylsilanes in a variety of organic synthetic routes and the improved methodologies of their preparation have turned organosilanes into important reagents for organic chemistry. This review discusses the recent employment of acylsilanes in organic synthesis and also effective methods for their preparation.
Keywords: organosilicon compounds, Brook rearrangement, carbonyl compounds, stereocontrol
Introduction
Acylsilanes (RCOSiR´3) are compounds that have the silicon directly attached to the carbonyl group, exhibiting unique chemical properties. The use of acylsilanes in organic synthesis has increased significantly over the last few years due to the discovery of valuable new reactions and the improvement of acylsilane synthesis methods. The substantial effect of the electronic properties and the bulk of trimethylsilyl group may be used to control the stereochemistry of reactions1. One of the well-established uses of acylsilanes in organic synthesis is as an aldehyde equivalent, in which a stereoselective nucleophilic attack on the carbonyl group, a to a chiral center, is followed by stereospecific replacement of the silyl group by hydrogen. Moreover, acylsilanes can be used as ester equivalents for chirality induction, since acylsilanes can be smoothly oxidized to esters. The enantioselective reduction and the cyclization reaction of acylsilanes appear to have substantial synthetic potential. In the first part of this review we present some physical properties of acylsilanes as well as some classical reactions involving organosilicon compounds. In the second part, we present some new work in this area as a complement to the previous reviews2-7 and finally, in the third part, useful methods of acylsilane synthesis.
Physical properties and classical reactions of the acylsilanes
In the organosilicon compounds class, the acylsilanes are compounds that present particular physical and chemical properties. The spectral data of acylsilanes are well described in the reviews by Brook2 and Page and co-workers3. The inductive effect of the silicon favors the polarization of the carbonyl group, which absorbs at a lower frequency than simple ketones in the infrared and ultraviolet spectra. In 13C NMR spectroscopy, the signals for the carbonyl carbon are quite different from the corresponding ketones, appearing at higher d values. The anisotropy effect and electronegativity differences also lead to higher d values in the 1H NMR spectra for hydrogens attached to the a-carbon of acylsilanes (except for a,b-unsaturated). Table 1 shows some examples of IR and NMR spectral data for acylsilanes. Another interesting characteristic of acylsilanes is the abnormally long Si-CO bond (1.926 Å), first observed by Trotter8 based on X-ray analysis, which can be compared to the analogous bond length in C-CO (1.51 Å)2 compounds. The same authors determined that the carbonyl bond length is essentially normal, despite the low vibrational frequency.
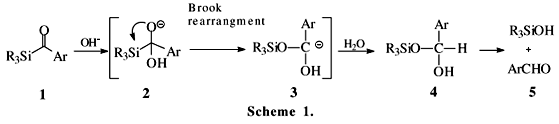
Studies have shown that acylsilanes in general behave as ordinary ketones. However, in some cases, these compounds have abnormal chemical behavior. Due to the intrinsic properties already mentioned, these compounds should undergo many unusual reactions. For example, in reactions of aroylsilanes with nucleophiles, the Brook rearrangement9 is very common. An example is the known hydrolysis of aroylsilanes 1 to the corresponding aldehydes 5, promoted by traces of OH- (Scheme 1).
The Brook rearrangement, after carbonyl addition of a nucleophilic reagent, is commonly observed in aroylsilanes due to the relative stabilization of the carbanion intermediate 3 by the aromatic ring. Two classical examples in this context are presented in Scheme 2, where the silyloxyalkene 6 is formed only when an anion-stabilizing group is attached to the carbonyl group2.

Brook investigated the great reactivity of acylsilanes towards nucleophilic addition9,10. His work is historically important because it led to a better knowledge of the reaction pathway. A nucleophilic attack of alkoxide ion on the silicon atom of acylsilane 7 was initially proposed for the formation of aldehyde 8 (Scheme 3, pathway a). Later, Brook proposed another competitive pathway, which involves a nucleophilic attack by the alkoxide ion at the carbonyl group of 7 (Scheme 3, pathway b). Using the optically active acylsilane (-)-9 in reactions with different alkoxide ions, 10 was reduced by LiAlH4 to give (-)-11 (Scheme 4). Although the optical purities of (-)-11 were observed to be dependent on the bulk of the alkoxide ion (EtO- 22% vs. t-BuO- 65%), in all reactions with chiral acylsilane (-)-9 the enantiomer (-)-11 was predominant, showing the retention of configuration at silicon. Therefore the bulkier alkoxides find it more difficult to attack at silicon and, consequently, the attack at the carbonyl group becomes relatively easier11.


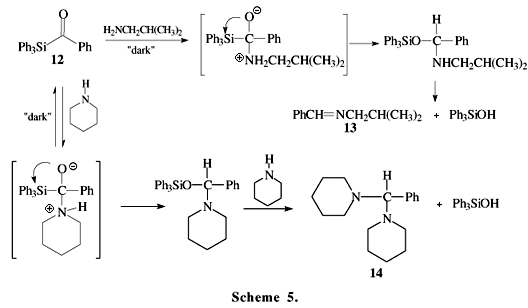
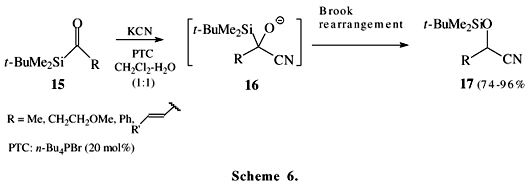
Schemes 5 and 6 show recent examples of acylsilane reactions involving the Brook rearrangement. The acylsilane 12 reacts with amines to form the imine 13 or aminals 1412. The acylsilane 15 reacts with cyanide anion under liquid-liquid (CH2Cl2-H2O) phase-transfer catalytic conditions to form O-silylated cyanohydrin 1713.
Novel work involving acylsilane chemistry
Herein some of the most important synthetic applications of acylsilanes will be presented, focusing especially on reports of the last ten years.
a. Stereocontrolled nucleophilic additions
The first study involving enantioselective addition to acylsilanes was reported by Mosher14 in which he used an optically active Grignard reagent to reduce benzoyl-triphenylsilane and benzoyltrimethylsilane in low enantiomeric excess. Due to the relative facility that the silyl moiety can be removed and replaced by hydrogen, generally by the action of fluoride ion, acylsilanes can be considered as aldehyde equivalents in nucleophilic additions. Addition of nucleophiles can occur with a high Cram selectivity when the chiral center in the acylsilane is on the a-carbon, as in 18 (Scheme 7), and even in some cases where the chiral center is on the b-carbon1, as in 19 (Scheme 8).


In general, the protiodesilylation (replacement of the R3Si moiety by H) occurs with a high level of stereoselectivity through the Brook rearrangement15.Two recent examples are shown in the Schemes 8 and 9 in which high stereocontrol was obtained in asymmetric induction in the syntheses of calcitriol lactone derivatives 2016 and b-aminoalcohols 21 and 2217. Scheme 10 illustrates a key step of Corey's total synthesis of pentacyclic triterpene 24 of the b-Amyrin family18. The addition of 2-propenyl-lithium to acylsilane 23, followed by a Brook rearrangement and coupling with allylic bromide, resulted in the Z-olefin in an overall yield of 82% with excellent stereoselectivity (> 95%).


Syn-a-alkoxy-(b-silyloxy)acylsilanes 25 undergo chelation-controlled addition reactions with vinyl and phenyl Grignard reagents affording all-syn triols 26 with diastereoselectivities up to >98:2 (Scheme 11)19,20.

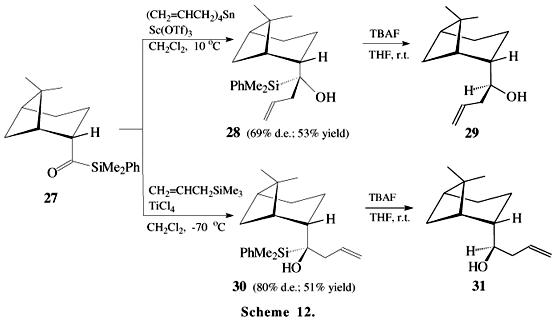
The allylation reaction of acylsilane 27 has been applied to a stereoselective synthesis of allyl myrtanols 29 and 31 (Scheme 12)21. In this case, the reaction of 27 with tetraallyltin/Sc(OTf)3 provides an asymmetric induction opposite to that observed by using allyltrimethylsilane/TiCl4. The configurations of 28 and 30 were tentatively assigned taking into account that the protiodesilylation generally proceeds with complete retention of configuration15,22.The B-allyl(diisopino-campheyl)-borane has been also used for asymmetric allylation of acylsilanes 32 (Scheme 13)23.

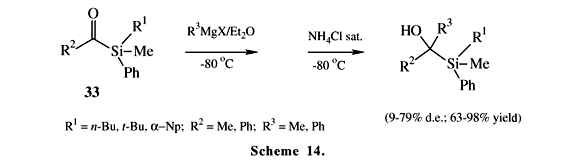
The addition of alkyl and phenyl lithium24,25 or Grignard reagents (Scheme 14)26 to acylsilanes 33 having a chiral center at silicon is also diastereoselective. High diastereo-selective excesses are obtained with Grignard reagents by means of a chelate-controlled reaction pathway involving intermediate 34 (Scheme 15)24. Cyclopropanediol monosilyl ethers 36 and 37 are obtained with good diastereoselectivity from reaction of benzoylsilane 35 with lithium enolates derived from methylketones (Scheme 16)27.


Ethynyl triphenylsilyl ketone 38 undergo stereospecific Michael addition with silylated nucleophiles28, dialkyl-cuprates29 and tributylstannyl-cuprate30 to afford only one double bond isomer of b-functionalized propenoylsilanes depending on the type of nucleophile used. An interesting application of these reactions is the preparation of a variety of a,b-unsaturated acylsilanes 40 by reaction of 38 with trimethylsilyl iodide yielding 39, which undergoes stereospecific palladium-catalyzed coupling with tin compounds (Scheme 17)31.

b. Stereocontrolled aldol reactions
Lithium enolates of propanoyl silanes 41 react with aryl and alkyl aldehydes to afford mainly syn-b-hydroxyacylsilanes 42, which can be converted into 43 as the major product (Scheme 18) in 31-68% overall yields32. While benzaldehyde gives a modest syn/anti ratio, isobutyraldehyde gives good diastereoselectivity (syn/anti > 20). Adehydes having a chiral center on the a-carbon react with 41, giving 44 in good diastereoselectivity.

Stereoselective intramolecular aldol reaction was performed with bis-acylsilanes under Lewis acid activation to give cis-b-hydroxyacylsilanes33.
c. Acylsilanes in radical reactions
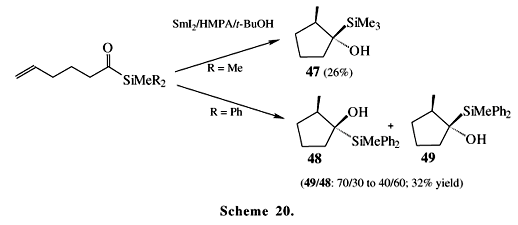
Lanthanoids, especially ytterbium and samarium, promote several kinds of reactions with alkanoyl and aroylsilanes. The mechanism of these reactions is always dependent on the metal and on the group attached to the carbonyl group (Scheme 19). An interesting point is the production of acetylene derivatives 46, which are probably formed by the addition of a silyl-radical 45 to another acylsilane molecule and involve two consecutive Brook rearrangements34. Other reactions such as intramolecular radical cyclization reactions of acylsilanes (Scheme 20), aldol reactions (Scheme 21) and pinacol couplings (Scheme 22) take place mediated by SmI235. The example presented in Scheme 20 shows an interesting cis-stereoselectivity observed for the cyclization of the acyltrimethylsilyl group (giving 47), while the diphenyl-methylsilyl group gave poor (48:49) stereoselectivity.


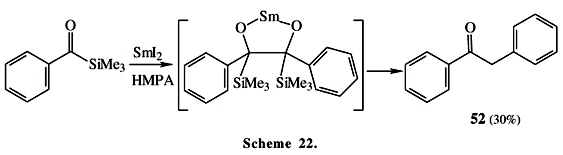
Trialkyltin radicals can promote intramolecular cyclization of acylsilanes 53-55 with alkyl, aryl and vinyl radicals, affording cyclopentyl silyl ethers 56 and 57 and enol silyl ether 58 (Scheme 23)36 by a mechanism involving a radical Brook rearrangement, as shown in the example outlined in the Scheme 2436c. An interesting application of the tandem cyclization-addition reaction of acylsilanes is the diastereoselective synthesis of endo bicyclic alcohol 60 from acylsilane 59 (Scheme 25)36a. On the other hand, bicyclic spiro-lactones 62 and 63 are obtained in the reaction of 61 with methyl acrylate and tributyltin hydride or CH2=C(CO2Et)CH2SnBu 3 (Scheme 26)37. For other examples see Tsai38, including tandem cyclizations with acylsilanes containing C=C or CºC groups attached at the silicon atom.




d. Cyclization reactions of acylsilanes
In addition to the examples cited above, many other cyclization reactions involving acylsilanes can be found in the literature. Carbonyl acylsilanes 64 provide furans 65 under milder conditions and in higher yields than the common cyclization reactions of dicarbonyl compounds. This advantage is derived from the high nucleophilicity of the carbonylic oxygen in acylsilanes, due to the contribution of the polarized resonance form II (Scheme 27)39. Scheme 28 presents examples of syntheses of disilylhydropyranes 68and disilylfurans 69 through a similar cyclization of 1,5-bis-acylsilanes 6640 and 1,4-bis-acylsilanes 6741 respectively, catalyzed by p-toluene-sulfonic acid (TsOH). The nucleophilicity of the carbonylic oxygen in acylsilane is also observed in the cyclization of g- and d-haloacylsilanes 70 affording 71 in reasonable to good yield (Scheme 29)42.



a,b-Unsaturated acylsilanes 72 combine with allenylsilanes 73 in presence of TiCl4 to produce [3+2] or [3+3] annulation products 74 and 75 in good yield. The course of the annulation reactions can be controlled to produce either five- or six-membered rings by controlling the reaction temperature or by using an appropriate trialkylsilyl group in 72 (Scheme 30)43, since the initially produced cyclopentene undergoes rearrangement at higher temperatures.

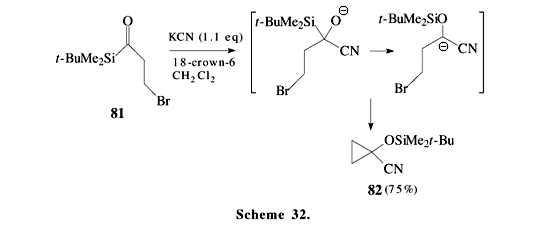
On the other hand, a,b-unsaturated acylsilanes 76 and lithium enolates of a,b-unsaturated methyl ketones 77 afford interesting [3+4]-annulation products 78 (Scheme 31)44. These stereospecific reactions were proposed to occur by a concerted anionic oxy-Cope rearrangement through cyclopropanediolate intermediate 80. The addition of cyanide anion13,45 to acylsilane 81 (or phenyllithium in an analogous reaction)46 produces a cyanohydrin which undergoes a Brook rearrangement, followed by an intramolecular alkylation to give cyclopropane 82 (Scheme 32).

e. Thioacylsilanes: preparation and synthetic applications
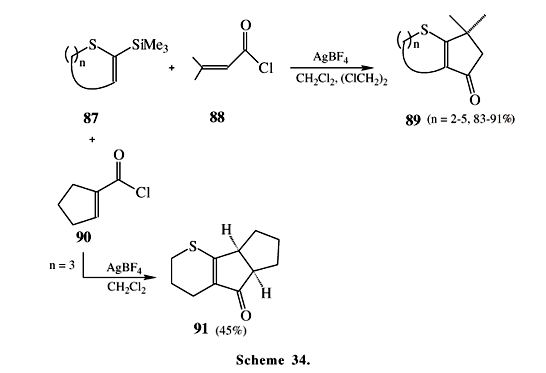
The replacement of the C=O with the C=S functionality in acylsilanes is possible by reacting acylsilanes such as 83 with H2S/HCl or by treatment with (Me3Si)2S under CoCl2 catalysis47. These highly reactive compounds are employed as unstable thioaldehyde equivalents and also to afford molecules containing the Si-C-S unit or to prepare sulfur heterocycles. Contrary to the thiocarbonyl analogues, the thioacylsilanes as 84 having an a-hydrogen bonded to the C=S group exist in the thioenol tautomeric form affording compounds Z-a-silyl vinyl mercaptans48, such as 85. These are interesting molecules because two functional groups with opposite polarization are bonded to a single double bond. An example of these reactions is concisely outlined in Scheme 33 where halothioacylsilanes 84 are cyclized in the presence of a base to provide 2-silyl-thiocycloalkyl-2-enes 86 in good yields49. These compounds react with acid chlorides, such as 88 and 90, in presence of a Lewis acid to give interesting bicyclic 89 or tricyclic 91 structures depending upon the nature of the alkyl chloride (Scheme 34)50. Scheme 35 presents some reactions of thioacylsilanes 92 containing the ferrocene moiety attached to the thiocarbonyl group51. Unlike ketones and common acylsilanes, which need long reaction periods and high temperatures for replacement of the C=O with the C=S functionality, acylsilanes containing ferrocene are converted readily into thioacylsilanes by Lawesson's52 reagent in high yields in a few minutes at room temperature.


f. Acylsilanes derived from natural products
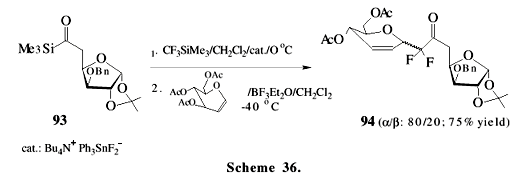
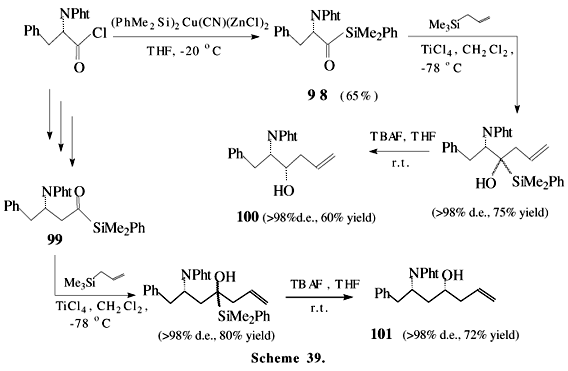
We have already described the application of acylsilanes for the synthesis of a pentacyclic triterpene, vitamin D3 metabolites and in aminoalcohol synthesis. However, the use of acylsilane moiety in natural product synthesis is still very restricted and few examples are found in the literature. Scheme 36 shows an "acylsilane-sugar" 93 as the starting material to prepare C-difluorosaccharide 94. Sugar chemistry may be a good option for the preparation of optically active silanes, little explored to date53. The proposed mechanism of this condensation of the saccharides is based on the highly reactive difluorosilyl enol ether 95, formed by the reaction of acylsilane with trifluoromethylsilane, followed by Brook rearrangement and the subsequent loss of fluoride ion, as outlined in Scheme 3754. The reaction of acylsilanes with trifluoromethyl-trimethylsilane has been utilized to afford alicyclic 2-fluoro-1,3-diketone55, 2,2-difluoro-1,5-diketone56 and "difluoro-ar-tumerone" 9757, a sesquiterpene derivative with antitumor activity, as shown in Scheme 38. The stereoselective synthesis of b- and g-amino alcohols 100 and 101, starting from a natural aminoacid and proceeding through the homochiral aminoacylsilanes 98 and 99, reveals the great potential of these compounds as chiral building blocks (Scheme 39)58.


g. Reactions of a-haloacylsilanes
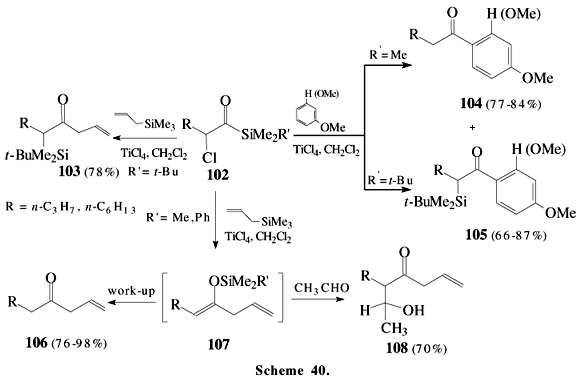
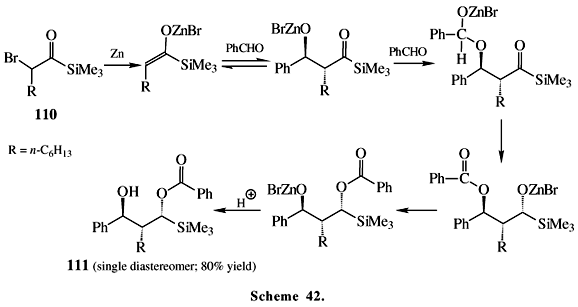
In spite of a-haloacylsilanes being little known, their reactions are very interesting due to a variety of products that they provide. Ketone enolates usually react with a-chloroacyltrimethylsilane under mild reaction conditions to give the corresponding 1,3-dicarbonyl compounds59. Titanium tetrachloride promotes coupling reactions of a-chloroacylsilanes 102 to afford a-silyl ketones 103, b,g-unsaturated ketones 106 and Friedel-Crafts type products 104 and 105 (Scheme 40). The intermediate acyl cation equivalent 109 was proposed to be responsible for the formation of 103 (Scheme 41). An interesting observation in these reactions is the migration of silyl group and subsequent loss of this moiety, which happens when R' = Me or Ph, but not t-butyl. Apparently, the formation of the intermediate silyl enol ether 107 is difficult when the silyl group is very bulky (R' = t-Bu). The presence of intermediate 107 was inferred from the aldol type products 10860 isolated upon addition of acetaldehyde to the reaction mixture. Another example involving these a-haloacylsilanes is presented in Scheme 42, where a-bromoacylsilane 110 reacts with zinc in a Reformatsky type reaction giving 111 with high diastereoselectivity61. The addition of triethylborane to a-iodoacylsilane provides boron enolate that reacts with aldehydes to give aldol types adducts with good diastereoselectivity in reasonable yields61.

h. Acylsilane oxidation
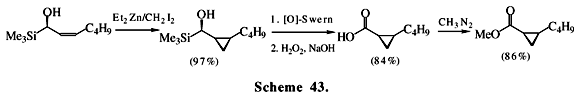
The acylsilanes have a much smaller oxidation potential than aldehydes and ketones due to the large interaction of the C-Si bond with the lone electron pair of the carbonyl oxygen62. This characteristic allows the acylsilanes to suffer oxidation to the corresponding carboxylic acids mediated by peroxides63, ozone64 and electrochemistry65. The oxidation of acylsilanes has been showing of great importance in organic synthesis, for example in chain homologation66 and as a precursor for chiral esters (Scheme 43)67. The most common method for oxidation of acylsilanes utilizes peroxides but the electrochemical method, although seldom applied, appears to be advantageous because it permits the direct preparation of acids, esters or nitrogen derivatives, depending upon the additive present during electrolysis. Recently, we introduced a new method for direct esterification of alkyl and aroylsilanes by means of iron (III) ion or nitric acid oxidation in dilute alcohol solution (Scheme 44)68.

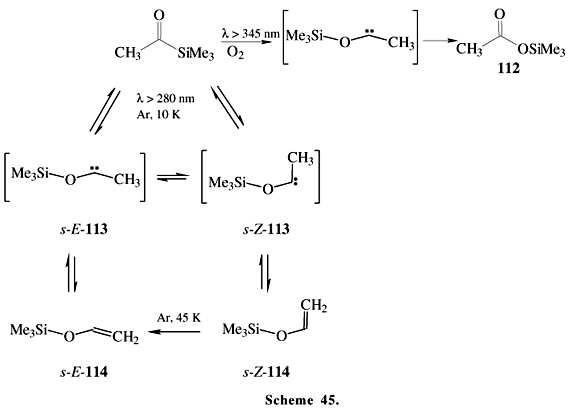
In this context, an important property of the acylsilanes is their oxidation through photoprocesses, affording compounds such as carboxylic acids (promoted by room light)69,70 or silylesters 112 and viniloxysilanes 114 as principal products (Scheme 45)71. Also see Brook72 for other examples of classical photolyses reactions of acylsilanes.
i. Enantioselective reduction of acylsilanes
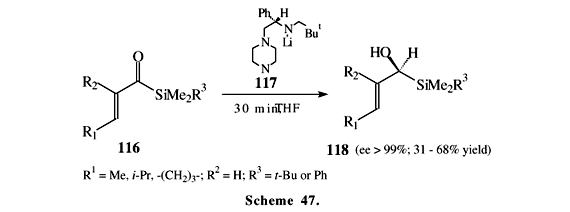
Chiral boranes have been used for enantioselective reduction of acylsilanes affording optically active alcohols (Scheme 46)73,74. Compound 115 was used as homochiral building block in the synthesis of (+)-sesbanimide A20. Scheme 47 shows an interesting reduction of a,b-unsaturated acylsilanes 116, which is mediated by the chiral lithium amide 117 affording alcohols 118 in excellent enantiomeric excesses75.

Because of the higher reactivity of the carbonyl group of acylsilanes compared to the analogous ketones, attributed mainly to the high polarization of the acylsilane carbonyl group, these compounds can react with weak nucleophiles. Therefore, these organosilanes are important substrates for bioreductions, from which the corresponding a-hydroxysilanes can be obtained with high enantiomeric excesses by using a variety of isolated enzymes or microorganisms76. Scheme 48 shows one of the first reports of this methodology, the acylsilane 119 being reduced by Trigonopsis variabilis 20 times faster than the analogue dimethylphenylpropanone77. Although Saccharomyces cerevisiae is one of the most commonly used micro-organisms in enantioselective reductions of ketones, it is usually inert toward substrates possessing steric hindrance78 or having electron-donating groups attached to the aromatic ring of the aroyl ketone79. On the other hand, aroylsilanes such as 121 were reduced by this microorganism, affording the corresponding a-hydroxy-silanes 120 and 122 with enantiomeric excesses varying from 43 to 88%, Scheme 4980. The disadvantage in using acylsilane bioreductions is the required lengthy period of reaction, which generally leads to the formation of by-products such as primary alcohols and carboxylic acids from C-Si bond cleavage. A radical oxidation of acylsilanes seems to be responsible for the formation of carboxylic acids81.


In Scheme 50 there is an interesting example of the bioreduction of a racemic acylsilane (±)-123 containing asymmetric silicon that was applied to afford chiral silyl compounds. Thus, acylsilane (-)-123, prepared by oxidation of (+)-124, was treated with phenyllithium, the asymmetric silyl group being a chiral inductor. After the Brook rearrangement promoted by KH, the silyl group was removed by tetrabutylammonium fluoride (TBAF), giving the enantiomerically enriched alcohol (R)-(+)-12725. Chiral secondary silyl alcohols may also suffer thermal rearrangements through their acylated derivatives followed by oxidative cleavage74,82 that occurs with high stereocontrol83, giving optically active alcohols.

Synthesis of acylsilanes
In 1957, Brook reported the first synthesis of an acylsilane, benzoyltriphenylsilane, which was accomplished by the reaction of triphenylsilylpotassium with benzoyl chloride in only 6% yield. The same compound was prepared in much better yield by hydrolysis of dihalide compound 128 (Scheme 51)84.

The biggest problem in the synthesis of acylsilanes is the instability of these compounds under many reaction conditions which may lead to C-Si bond cleavage. In this review, some of the methodologies developed for acylsilanes synthesis will be introduced concisely. A useful summary is presented in the Chart, showing that some of the common organic functional groups may be used for the preparation of acylsilanes.

a. From a-silyl alcohols2,4
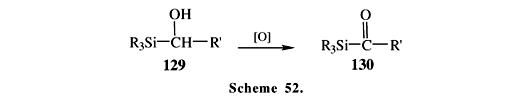
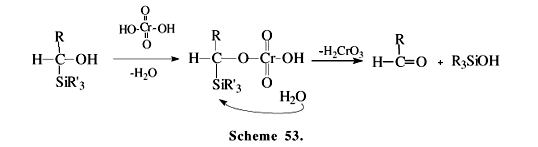
a-Silyl alcohols can be prepared by several methods, such as the condensation of trialkylsilyl anions with aldehydes85 and the transmetalation of trialkylstannanes followed by a reverse Brook rearrangement86. The oxidation of a-silyl alcohol 129 with ordinary oxidizing reagents like permanganate and chromic acid leads to acylsilanes 130 (Scheme 52). However, this route has several limitations since Si-C bond cleavage may compete (Scheme 53)2, and the products may suffer over-oxidation to carboxylic acids (Schemes 43 and 44). Very mild conditions, such as those present in Swern oxidation, are the most indicated options. In the example showed in Scheme 5487,3, the "reverse Brook rearrangement" (131®132), followed by a mild oxidation, is employed for the synthesis of a,b-unsaturated acylsilanes 133. Silylalcohol 135, prepared by nucleophilic opening of epoxide 134, was oxidized under extremely mild condition by the use of the Dess-Martin reagent, giving acylsilanes 136 in good to excellent overall yields (Scheme 55)88. Recently, we have found that potassium permanganate supported onto alumina transforms silylalcohols to aroylsilanes in good yields, without significant Si-C bond cleavage or over-oxidation to carboxylic acid89.


b. From masked aldehydes
The addition of silyllithium reagents to aldehydes gives a-silyl alcohols, which may be oxidised to acylsilanes as commented above. However, aldehydes 137 are more commonly converted into acylsilanes 139 by the dithiane route (the umpolung methodology90, Scheme 56). The hydrolysis of 2-silyl-1,3-dithianes 138 was first investigated in simultaneous work by Brook91 and Corey92, and it is one of the most useful methodologies for acylsilanes synthesis. The great advantage of this method is the variety of compounds that can be prepared, including aroylsilanes, alkanoylsilanes, and functionalized acylsilanes. In general, the first and second steps (Scheme 56) afford products in high yields, but the hydrolysis step may be problematic. Although there are many procedures for the regeneration of the masked carbonyl group in dithianyl compounds93, the most frequently applied reagents for 2-silyldithianes 138 are mercury salts, the oldest methodology for hydrolysis. Generally, the hydrolysis with mercuric chloride is very slow, thus the product aroylsilanes may suffer degradation, as mentioned earlier. Other methods have been applied for the regeneration of the masked carbonyl group, giving acylsilanes in good yields. Among these, we may mention treatment with methyl iodide41, chloroamine-T33, I2/CaCO394, anodic oxidation95 and oxidative hydrolysis mediated by N-bromosuccinimide (NBS)96.

The benzotriazole derivatives 140 present a similar approach to the dithiane methodology, offering advantage in the hydrolysis step. The hydrolysis of intermediate 141 occurs in situ and under mild conditions, giving the desired aroyl, heteroaroyl, alkenoyl and alkynoylsilanes 142 in excellent yields (Scheme 57)97.

c. From esters and amides
The synthesis of acylsilanes by reductive silylation is well known and proceeds by the reaction of esters with silyl-Grignard derivatives, forming silylacetals 143 (Scheme 58)98. This method generally gives poor yields and hence has been seldom employed. One-pot synthesis of aroylsilanes based on reductive silylation of methylbenzoates using Mg/I2/chlorosilane/NMP (1-methyl-2-pyrrolidinone) affords aroylsilanes in moderate yields99.

Compounds containing lithium attached to the silicon are extremely important reagents in organosilane chemistry. Dimethylphenylsilyllithium (144) is probably the most useful silyllithium for synthesis, due to the aryl group that gives good anion stability and to the fact that this reagent can be readily prepared from the corresponding chlorodimethylphenylsilane by its reaction with lithium in THF100. On the other hand, trimethylsilyllithium is readily obtained by the reaction of hexamethyldisilane with methyllithium101. These compounds react with esters or amides to afford acylsilanes (Scheme 59)102. Amides appear to be more useful, giving better yields than the traditional reaction with esters, which require much lower temperatures.

In general, the reaction of silyllithium with esters affords disilylalcohols as undesirable by-products due to double nucleophilic attack at the carbonyl group. However, these alcohols, such as 145, have been oxidized by PDC (pyridinium dichromate)102, tert-butyl hypochlorite103 and lead tetraacetate104 (Scheme 60) to the corresponding acylsilanes. This latter method was recently reported, and involves a "radical Brook rearrangement" providing acylsilanes in good yield after treatment with silica gel.

d. From S-2-pyridyl esters
S-2-Pyridyl esters 146 react very smoothly with Al(SiMe3)3 in the presence of CuCN to afford acylsilanes in good to excellent yields (Scheme 61). This method may be applied to substrates having various groups such as alkoxyl, acetal, ester, or an isolated double bond. Chiral centers a to the carbonyl group are not epimerized under the reaction conditions105.

e. From acid chloride
Treatment of silyllithium with acid chloride gives acylsilanes. However, this procedure is not general due to the complex reaction mixtures that it provides. On the other hand, lithium silylcuprates like 148 react with a variety of acid chlorides, giving acylsilanes with good yields, offering advantages over the silyllithium methodology since fewer by-products are formed106. These cuprates are traditionally obtained from the reaction of an alkylsilyllithium with CuCN or CuI107. A limitation of this methodology is that high order cuprates are very reactive towards a variety of functional groups. The mixed Cu-Zn complex 147 is less reactive than ordinary cuprates, and therefore this type of complex has been applied to the synthesis of acylsilanes containing cyano, halo, ester and other groups (Scheme 62)6,108.

Yamamoto and co-workers109 prepared aroylsilanes by reaction of disilanes (compounds with Si-Si bond) with benzoyl chloride under palladium catalysis. However this method is not suitable for aliphatic acylsilanes, giving low yields of products. The methodology presented in Scheme 63 is a good alternative, providing both aroyl and alkanoylsilanes by reacting the acid chlorides 149 with the polarised Si-Sn bond of 150 (weaker than the Si-Si bond in the disilanes)110.

f. From other organic functionalities
The application of a,b-unsaturated acylsilanes as building blocks for organic synthesis is well known in such important reactions as the Diels-Alder one111, thermal rearrangements and cyclizations, as commented above. A common method for alkenoylsilane synthesis goes through the allenylsilanes 151 and is known as Reich's procedure (Scheme 64)113. Scheme 65 summarizes another method for the preparation of a-substituted-a,b-unsaturated acylsilanes 155 from silylpropargyl derivative 152 in a methodology involving silyloxy allene formation, a "reverse Brook rearrangement" (153®154) followed by the addition of an aldehyde.


Several methods for the preparation of haloacylsilanes are reported in the literature, such as bromination of silyl enol ethers115 and halogenation of alkyl enol ethers116 with N-bromosuccinimide (NBS) or N-chlorosuccinimide (NCS). The reaction of ketones with silyl carbanion 156, which is obtained from an ethereal solution of t-BuMe2SiCHBr2 and lithium diisopropylamide, provides silyloxiranes 157 which undergo rearrangement, yielding a-bromoacylsilanes 158 in reasonable yields (Scheme 66)117. This method is a recent example of an a-bromoacylsilane preparation, where the formation of 2-silyloxiranes 157 is proposed by the authors, based on a known method for synthesis of a-iodo, a-fluoro, a-bromo and a-chloroacylsilanes 160 from silyloxiranes 159 (Scheme 67)61.


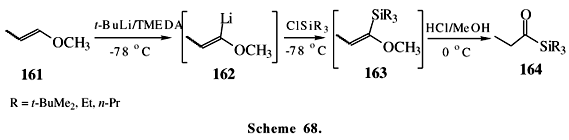
Acylsilane 164 could be prepared in a one-pot procedure from vinyl ether 161 by the reaction of intermediate 162 with trialkyl silyl chloride in pentane at 78 oC, followed by mild acid hydrolysis of 163 (Scheme 68)32,118. Starting with allylic alcohol 165, acylsilane 23 was prepared in a one pot procedure (Scheme 69)18.This acylsilane was used in the Corey's total synthesis of triterpenes as presented earlier in Scheme 10.

Conclusion
In this review we have resumed the most significant articles, published in the last ten years, involving acylsilanes, both from the point of view of their preparation and their application as versatile tools in organic syntheses. The research in this area has increased significantly over the last few years due to the discovery of valuable new reactions and the improvement of acylsilane synthesis methods. It is possible to visualise that in the near future the acylsilanes will be a well-known tool for the use of organic synthetic chemists.
References
1. Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063.
2. Brook, A. G. In Adv. Organomet. Chem., Stone, F. G. A.; West, R. Eds.; Academic. Press, N.Y, 1968, 7, p 95.
3. Page, P. C. B.; Klair, S. S.; Rosenthal, S. Chem. Soc. Rev. 1990, 19, 147.
4. Ricci, A.; Degl'Innocenti, A. Synthesis 1989, 647.
5. Cirillo, P. F.; Panek, J. S. Org. Prep. Proced. Int.1992, 24, 555.
6. a) Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A. Gazz. Chim. Ital. 1997, 127, 619;
b) Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A. J. Organomet. Chem. 1998, 567, 181.
7. Nájera, C. Yus, M. Org. Prep. Proced. Int. 1995, 27, 383.
8. Chieh, P. C.; Trotter, J. J. Chem. Soc. 1969, 1778.
9. (a) Brook, A. G.; Warner, C. M.; McGriskin, M. J. Am. Chem. Soc. 1959, 81, 981. (b) Brook, A. G. Acc. Chem. Res. 1974, 7, 77. (c) Brook, A. G.; Bassindale, A. R. In: Rearrangements in Ground and Exited States; de Mayo, P., Ed; Academic Press, New York, 1980.
10. (a) Brook, A. G. J. Org. Chem. 1960, 25, 1072. (b) Brook, A. G.; Schwartz, N. V. J. Org. Chem. 1962, 27, 2311.
11. Brook, A. G.; Vandersar, T. J. D.; Limburg, W. Can. J. Chem. 1978, 56, 2758.
12. Brook, A. G.; Yu, Z. F. Organometallics 2000, 19, 1859.
13. Takeda, K.; Ohnishi, Y. Tetrahedron Lett. 2000, 41, 4169.
14. Biernbaum, M. S.; Mosher, H. S. J. Org. Chem. 1971, 36, 3169.
15. Hudrlik, P. F.; Hudrilik, A. M.; Kulkarni, A. K. J. Am. Chem. Soc. 1982, 104, 6809.
16. Nakada, M.; Urano, Y.; Kobayashi, S.; Ohno, M. Tetrahedron Lett. 1994, 35, 741.
17. (a) Bonini B. F.; Franchini, M. C.; Laboroi, F.; Mazzanti, G.; Ricci, A.; Varchi, G. J. Org. Chem. 1999, 64, 8008. (b) Bonini B. F.; Franchini, M. C.; Fochi, M.; Gawronski, J.; Mazzanti, G.; Ricci, A.; Varchi, G. Eur. J. Org. Chem. 1999, 437.
18. Huang, A. X.; Xiong, Z.; Corey, E. J. J. Am. Chem. Soc. 1999, 121, 9999.
19. Cirillo, P. F.; Panek, J. S. J. Org. Chem. 1990, 55, 6071.
20. Cirillo, P. F.; Panek, J. S. J. Org. Chem. 1994, 59, 3055.
21. Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Nanni, C.; Ricci, A. Tetrahedron Lett. 1998, 39, 6737.
22. Nakada, M.; Urano, Y.; Kobayashi, S.; Ohno, M. J. Am. Chem. Soc. 1988, 110, 4826.
23. Buynak, J. D.; Geng, B.; Uang, S.; Strikcland, J. B. Tetrahedron Lett. 1994, 35, 985.
24. Chapeaurouge, A.; Bienz, S. Helv. Chim. Acta 1993, 76, 1876.
25. Huber, P.; Bratovanov, S.; Bienz, S.; Syldatk, C.; Pietzsch, M. Tetrahedron: Asymmetry 1996, 7, 69.
26. Bonini, B. F.; Masiero, S.; Mazzanti, G.; Zani, P. Tetrahedron Lett. 1991, 32, 6801.
27. Takeda, K.; Nakatani, J.; Nakamura, H.; Sako, K.; Yoshii, E.; Yamaguchi, K. Synlett, 1993, 841.
28. Degl'Innocenti, A.; Capperucci, A.; Reginato, G.; Mordini, A.; Ricci, A. Tetrahedron Lett. 1992, 33, 1507.
29. Degl'Innocenti, A.; Stucchi, E.; Capperucci, A.; Mordini, A.; Reginato, G.; Ricci, A. Synlett 1992, 329.
30. Degl'Innocenti, A.; Stucchi, E.; Capperucci, A.; Mordini, A.; Reginato, G.; Ricci, A. Synlett 1992, 332.
31. Degl'Innocenti, A.; Capperucci, A.; Bartoletti, L.; Mordini, A.; Reginato, G. Tetrahedron Lett. 1994, 35, 2081. See reference 30 for other examples.
32. Schinzer, D. Synthesis, 1989, 179.
33. Bouillon, J. P.; Portella, C. Eur. J. Org. Chem. 1999, 1571.
34. Taniguchi, Y.; Fujii, N.; Takaki, K.; Fujiwara, Y. Appl. Organomet. Chem. 1995, 9, 377.
35. Chuang, T. H.; Fang, J. M.; Jiaang, W. T.; Tsai, Y. M. J. Org. Chem. 1996, 61, 1794.
36. (a) Tsai, Y. M.; Tang, K. H.; Jiaang, W. T. Tetrahedron Lett. 1993, 34, 1303; (b) Chang, S. Y.; Jiaang, W. T.; Cherng, C. D.; Tang, K. H.; Huang, C. H.; Tsai, Y. M. J. Org. Chem. 1997, 62, 9089; (c) Jiaang, W. T.; Lin, H. C.; Tang, K. H.; Chang, L. B.; Tsai, Y. M. J. Org. Chem. 1999, 64, 618.
37. (a) Giese, B.; Angew. Chem. Int. Ed. Engl. 1985, 24, 553; (b) Curran, D. P.; Jiaang, W. T.; Palovich, M.; Tsai, Y. M. Synlett 1993, 403.
38. Tsai, Y. M.; Tang, K. H.; Jiaang, W. T. Tetrahedron Lett. 1996, 37, 7767.
39. Siedem, C. S.; Molander, G. A. J. Org. Chem. 1996, 61, 1140.
40. Saleur, D.; Bouillon, J. P.; Portella, C. Tetrahedron Lett. 1999, 40, 1885.
41. Saleur, D.; Bouillon, J. P.; Portella, C. Tetrahedron Lett. 2000, 41, 321.
42. Tsai, Y. M.; Cherng, C. D.; Nieh, H. C.; Sieh, J. A. Tetrahedron 1999, 55, 14587.
43. Danheiser, R. L.; Fink, D. M. Tetrahedron Lett. 1985, 26, 2513.
44. Takeda, K.; Nakajima, A.; Takeda, M.; Okamoto, Y.; Sato, T.; Yoshii, E.; Koizumi, T.; Shiro, M. J. Am. Chem. Soc. 1998, 120, 4947.
45. (a) Takeda, K.; Ohtani, Y. Org. Lett. 1999, 1, 677. (b) Takeda, K.; Tanaka, T. Synlett 1999, 705.
46. Reich, H.; Holtan, R. C.; Bolm, C. J. Am. Chem. Soc. 1990, 112, 5609.
47. (a) Barbaro, G.; Battaglia, A.; Giorgianni, P.; Maccagnani, G.; Macciantelli, D.; Bonini, B. F.; Mazzanti, G.; Zani, P. J. Chem. Soc., Perkin Trans. 1 1986, 381. (b) Capperucci, A.; Degl'Innocenti, A.; Ricci, A.; Reginato, G. J. Org. Chem. 1989, 54, 19.
48. Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Peri, F.; Ricci, A. J. Chem. Soc., Perkin Trans. 1 1996, 2803.
49. (a) Bonini, B. F.; Franchini, M. C.; Mazzanti, G.; Ricci, A.; Fauzza, L. R.; Zani, P. Tetrahedron Lett. 1994, 35, 9227. (b) Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A. Tetrahedron 1996, 52, 4803.
50. Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A. Tetrahedron 1997, 53, 7897.
51. Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A.; Varchi, G. Tetrahedron Lett. 1999, 40, 6473.
52. Brillon, D. Sulfur Rep. 1992, 12, 297.
53. (a) Royon, R. P.; Portella, C. Tetrahedron Lett. 1996, 37, 6113. (b) Brigaud, T.; Lefebvre, O.; Royon, R. P.; Portella, C. Tetrahedron Lett. 1996, 37, 6115.
54. Brigaud, T.; Doussot, P.; Portella, C. J. Chem. Soc. Chem. Commun. 1994, 18, 2117.
55. Saleur, D.; Brigaud, T.; Bouillon, J. P.; Portella, C. Synlett 1999, 432.
56. Lefebvre, O.; Brigaud, T.; Portella, C. Tetrahedron 1998, 54, 5939.
57. Lefebvre, O.; Brigaud, T.; Portella, C. Tetrahedron 1999, 55, 7233.
58. Bonini, B. F.; Franchini, M. C.; Mazzanti, G.; Ricci, A.; Sala, M. J. Org. Chem. 1996, 61, 7242.
59. Kuwajima, I.; Matsumoto, K. Tetrahedron Lett. 1979, 4095.
60. Horiuchi, Y.; Oshima, K.; Utimoto, K. J. Org. Chem. 1996, 61, 4483.
61. Horiuchi, Y.; Taniguchi, M.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1995, 36, 5353.
62. Mochida, K.; Okui, S.; Ichikawa, K.; Tsuchiya, T.; Yamamoto, K. Chem. Lett. 1986, 805.
63. Miller, J. A.; Zweifel, G. J. Am. Chem. Soc. 1981, 103, 6217.
64. Linderman, R. J.; Chen, K. Tetrahedron Lett. 1992, 33, 6767.
65. Yohida, J.; Matsunagua, S.; Isoe, S. Tetrahedron Lett. 1989, 30, 5293.
66. Salzmann, T. N.; Ratcliffe, R. W.; Christensen, B. G.; Bouffard, F. A. J. Am. Chem. Soc. 1980, 102, 6161.
67. Sakaguchi, K.; Mano, H.; Ohfune, Y. Tetrahedron Lett. 1998, 39, 4311.
68. Patrocínio, A. F.; Moran, P. J. S. Synth. Commun. 2000, 30, 1419.
69. Brook, A. G.; Pierce, J. B. Abst. 149th Meeting Am. Chem. Soc. Detroit, 1965, p.2P.
70. Brook, A. G. Acc. Chem. Res. 1973, 6, 77.
71. Trommer, M.; Sander, W. Organometallics 1996, 15, 189.
72. Brook, A. G.; Pierce, J. B.; Duff, J. M. Can. J. Chem. 1975, 53, 2875.
73. (a) Soderquist, J. A.; Anderson, C. L.; Miranda, E. I.; Rivera, I.; Kabalka, G. W. Tetrahedron Lett. 1990, 31, 4677. (b) Buynak, J. D.; Geng, B.; Uang, S.; Strickland, J. B. Tetrahedron Lett. 1994, 35, 985.
74. Buynak, J. D.; Strikcland, J. B.; Lamb, G. W.; Khasnis, D.; Modi, S.; Williams, D.; Zhang, H. J. Org. Chem. 1991, 56, 7076.
75. Takeda, K.; Ohnishi, Y.; Koizumi, T. Org. Lett. 1999, 1, 237.
76. (a) Tacke, R.; Linoh, H.; Stumpf, B.; Abraham, W. R.; Kieslisch, K.; Ernest, L. Z. Naturforch 1983, 38b, 616. (b) Syldatk, C.; Stoffregen, A.; Wuttke, F.; Tacke, R. Biotech. Lett. 1988, 10, 731. (c) Fischer, L.; Wagner, S. A.; Tacke, R. Appl. Microbiol. Biotechnol. 1995, 42, 671. (d) Tacke, R.; Wagner, S.A.; Brakmann, S.; Wuttke, F. J. Organomet. Chem. 1993, 458, 13.
77. Tacke, R.; Linoh, H. In The Chemistry of Organic Silicon Compounds; Patai, S.; Rappoport, Z., Eds.; John Wiley & Sons Ltd., New York 1989, chapter 18.
78. (a) Csuk, R.; Glanzer, B.I. Chem. Rev. 1991, 91, 49. (b) Servis, S. Synthesis 1990, 1.
79. (a) Mitteilung, K.; Eichberger, G.; Faber, K.; Griengl, H. Monatsch. Chem. 1985, 116, 1233. (b) Deardorff, D. R.; Myles, D. C.; Macferrin, K. D. Tetrahedron Lett. 1985, 26, 5615. (c) Wendhausen, R.; Moran, P. J. S.; Joekes, I.; Rodrigues, J. A. R. J. Mol. Catal. B: Enzym. 1998, 5, 57.
80. Patrocínio, A. F.; Corrêa Jr., I. R.; Moran, P. J. S. J. Chem. Soc. Perkin Trans. 1 1999, 3133.
81. Patrocínio, A. F.; Moran, P. J. S. J. Chem. Res. (S) 2000, 404.
82. (a) Bassindale, A. R.; Brook, A. G.; Jones, P. F.; Lennon, J. M. Can. J. Chem. 1975, 53, 332. (b) Buynak, J. D.; Strikcland, J. B.; Hurd, T.; Pan, A. J. Chem. Soc. Chem. Commun. 1989, 89.
83. Tamao, K.; Kakui, T.; Akita, M.; Iwahara, T.; Kanatani, R.; Yoshida, J.; Kumada, M. Tetrahedron 1983, 39, 983.
84. Brook, A. G. J. Am. Chem. Soc. 1957, 79, 4373.
85. (a) Mori, A.; Fujita, A.; Ikegashira, K.; Nishihara, Y.; Hiyama, T. Synlett 1997, 693. (b) Wilson, S. R.; Hague, M. S.; Misra, R. N. J. Org. Chem. 1982, 47, 747. (c) Barretm, A. G. M.; Hill, J. M.; Wallace, E. M.; Flygare, J. A. Synlett 1991, 764. (d) Hudrlik, P. F.; Abdallah, Y. M.; Kulkarni, A. K.; Hudrlik, A. M. J. Org. Chem. 1992, 57, 6552. (e) Hiyama, T.; Obayashi, M. J. Org. Chem. 1983, 48, 912.
86. (a) Linderman, R. J.; Ghannam, A. J. Am. Chem. Soc. 1990, 112, 2392. (b) Linderman, R. J.; Chen, K. Tetrahedron Lett. 1992, 33, 6767.
87. Danheiser, R. L.; Fink, D. M.; Okano, K.; Tsai, Y. M.; Szczepanski, S. W. J. Org. Chem. 1985, 50, 5393.
88. Lipshutz, B. H.; Lindsley, C.; Susfalk, R.; Gross, T. Tetrahedron Lett. 1994, 35, 8999.
89. Patrocínio, A. F.; Moran, P. J. S. Synth. Commun., in press.
90. Corey, E. J.; Seebach, D. Angew. Chem., Int. Ed. 1965, 4, 1075.
91. Brook, A. G.; Duff, J. M.; Jones, P. F.; Davis, N. R. J. Am. Chem. Soc. 1967, 89, 431.
92. Corey, E. J.; Seebach, D.; Freedman, R. J. Am. Chem. Soc. 1967, 89, 434.
93. Gröbel, B. T.; Seebach, D. Synthesis 1977, 357.
94. Bouillon, J. P.; Portella, C. Tetrahedron Lett. 1997, 38, 6595.
95. Suda, K.; Watanabe, J.; Takanami, T. Tetrahedron Lett. 1992, 33, 1355.
96. Patrocínio, A. F.; Moran, P. J. S. J. Orgamet. Chem. 2000, 603, 220.
97. Katritzky, A. R.; Wang, Z.; Lang, H. Organometallics 1996, 15, 486.
98. Picard, J. P.; Calas, R.; Dunoguès, J.; Duffaut, N.; Gerval, J.; Lapouyade, P. J. Org. Chem. 1979, 44, 420.
99. Tongco, E. C.; Wang, Q.; Prakash, G. K. S. Synth. Commun. 1997, 27, 2117.
100. Fleming, I.; Roberts, R. S.; Smith, S. C. Tetrahedron Lett. 1996, 37, 9395.
101. Still, W. C. J. Org. Chem. 1976, 41, 3063.
102. Fleming, I.; Ghosh, U. J. Chem. Soc., Perkin Trans. 1 1994, 257.
103. Kuwajima, I.; Abe, T.; Minami, N. Chem. Lett. 1976, 993.
104. Paredes, M. D.; Alonso, R. Tetrahedron Lett. 1999, 40, 3973.
105. Nakada, M.; Nakamura, S.; Kobayashi, S.; Ohno, M. Tetrahedron Lett. 1991, 32, 4929.
106. Capperucci, A.; Degl'Innocenti, A.; Faggi, C.; Ricci, A. J. Org. Chem. 1988, 53, 3612.
107. (a) Fleming, I.; Marchi Jr., D. Synthesis 1981, 560. (b) Ager, D.; Fleming, I.; Patel, S. K. J. Chem. Soc., Perkin Trans. 1 1981, 2521.
108. Bonini, B.F.; Franchini, M.C.; Mazzanti, G.; Passamonti, U.; Ricci, A.; Zani, P. Synthesis 1995, 92.
109. Yamamoto, K.; Suzuki, S.; Tsuji, J. Tetrahedron Lett. 1980, 21, 1653.
110. Geng, F.; Maleczka Jr., R. E. Tetrahedron Lett. 1999, 40, 3113.
111. Reich, H. J.; Kelly, M. S.; Olson, R. E.; Holtan, R. C. Tetrahedron 1983, 39, 949.
112. Brook, A. G.; Ionkin, A.; Lough, A. J. Organometallics 1996, 15, 1275.
113. Reich, H. J.; Kelly, M. J. J. Am. Chem. Soc. 1982, 104, 1119.
114. (a) Tius, M. A.; Hu, H. Tetrahedron Lett. 1998, 39, 5937. (b) Stergiades, I. A.; Tius, M. A. J. Org. Chem. 1999, 64, 7547.
115. Kuwajima, I.; Abe, T.; Minami, N. Chem. Lett. 1976, 993.
116. Nowick, J. S.; Danheiser, R. L. Tetrahedron 1988, 44, 4113.
117. Shinokubo, H.; Oshima, K.; Utimoto, K. Tetrahedron 1996, 52, 14533.
118. Soderquist, J. A.; Hassner, A. J. Am. Chem. Soc. 1980, 102, 1577.
Received: November 14, 2000
Published on the web: February 10, 2001
FAPESP helped in meeting the publication costs of this article.
- 1. Fleming, I.; Barbero, A.; Walter, D. Chem. Rev 1997, 97, 2063.
- 2. Brook, A. G. In Adv. Organomet. Chem., Stone, F. G. A.; West, R. Eds.; Academic. Press, N.Y, 1968, 7, p 95.
- 3. Page, P. C. B.; Klair, S. S.; Rosenthal, S. Chem. Soc. Rev 1990, 19, 147.
- 4. Ricci, A.; Degl'Innocenti, A. Synthesis 1989, 647.
- 5. Cirillo, P. F.; Panek, J. S. Org. Prep. Proced. Int.1992, 24, 555.
- 6. a) Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A. Gazz. Chim. Ital. 1997, 127, 619;
- b) Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A. J. Organomet. Chem. 1998, 567, 181.
- 7. Nájera, C. Yus, M. Org. Prep. Proced. Int. 1995, 27, 383.
- 8. Chieh, P. C.; Trotter, J. J. Chem. Soc 1969, 1778.
- 9. (a) Brook, A. G.; Warner, C. M.; McGriskin, M. J. Am. Chem. Soc. 1959, 81, 981.
- (b) Brook, A. G. Acc. Chem. Res 1974, 7, 77.
- (c) Brook, A. G.; Bassindale, A. R. In: Rearrangements in Ground and Exited States; de Mayo, P., Ed; Academic Press, New York, 1980
- 10. (a) Brook, A. G. J. Org. Chem. 1960, 25, 1072.
- (b) Brook, A. G.; Schwartz, N. V. J. Org. Chem 1962, 27, 2311.
- 11. Brook, A. G.; Vandersar, T. J. D.; Limburg, W. Can. J. Chem. 1978, 56, 2758.
- 12. Brook, A. G.; Yu, Z. F. Organometallics 2000, 19, 1859.
- 13. Takeda, K.; Ohnishi, Y. Tetrahedron Lett. 2000, 41, 4169.
- 14. Biernbaum, M. S.; Mosher, H. S. J. Org. Chem. 1971, 36, 3169.
- 15. Hudrlik, P. F.; Hudrilik, A. M.; Kulkarni, A. K. J. Am. Chem. Soc 1982, 104, 6809.
- 16. Nakada, M.; Urano, Y.; Kobayashi, S.; Ohno, M. Tetrahedron Lett 1994, 35, 741.
- 17. (a) Bonini B. F.; Franchini, M. C.; Laboroi, F.; Mazzanti, G.; Ricci, A.; Varchi, G. J. Org. Chem 1999, 64, 8008.
- (b) Bonini B. F.; Franchini, M. C.; Fochi, M.; Gawronski, J.; Mazzanti, G.; Ricci, A.; Varchi, G. Eur. J. Org. Chem. 1999, 437.
- 18. Huang, A. X.; Xiong, Z.; Corey, E. J. J. Am. Chem. Soc 1999, 121, 9999.
- 19. Cirillo, P. F.; Panek, J. S. J. Org. Chem. 1990, 55, 6071.
- 20. Cirillo, P. F.; Panek, J. S. J. Org. Chem. 1994, 59, 3055.
- 21. Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Nanni, C.; Ricci, A. Tetrahedron Lett 1998, 39, 6737.
- 22. Nakada, M.; Urano, Y.; Kobayashi, S.; Ohno, M. J. Am. Chem. Soc. 1988, 110, 4826.
- 23. Buynak, J. D.; Geng, B.; Uang, S.; Strikcland, J. B. Tetrahedron Lett 1994, 35, 985.
- 24. Chapeaurouge, A.; Bienz, S. Helv. Chim. Acta 1993, 76, 1876.
- 25. Huber, P.; Bratovanov, S.; Bienz, S.; Syldatk, C.; Pietzsch, M. Tetrahedron: Asymmetry 1996, 7, 69.
- 26. Bonini, B. F.; Masiero, S.; Mazzanti, G.; Zani, P. Tetrahedron Lett 1991, 32, 6801.
- 27. Takeda, K.; Nakatani, J.; Nakamura, H.; Sako, K.; Yoshii, E.; Yamaguchi, K. Synlett, 1993, 841.
- 28. Degl'Innocenti, A.; Capperucci, A.; Reginato, G.; Mordini, A.; Ricci, A. Tetrahedron Lett. 1992, 33, 1507.
- 29. Degl'Innocenti, A.; Stucchi, E.; Capperucci, A.; Mordini, A.; Reginato, G.; Ricci, A. Synlett 1992, 329.
- 30. Degl'Innocenti, A.; Stucchi, E.; Capperucci, A.; Mordini, A.; Reginato, G.; Ricci, A. Synlett 1992, 332.
- 31. Degl'Innocenti, A.; Capperucci, A.; Bartoletti, L.; Mordini, A.; Reginato, G. Tetrahedron Lett. 1994, 35, 2081. See reference 30 for other examples.
- 32. Schinzer, D. Synthesis, 1989, 179.
- 33. Bouillon, J. P.; Portella, C. Eur. J. Org. Chem. 1999, 1571.
- 34. Taniguchi, Y.; Fujii, N.; Takaki, K.; Fujiwara, Y. Appl. Organomet. Chem 1995, 9, 377.
- 35. Chuang, T. H.; Fang, J. M.; Jiaang, W. T.; Tsai, Y. M. J. Org. Chem 1996, 61, 1794.
- 36. (a) Tsai, Y. M.; Tang, K. H.; Jiaang, W. T. Tetrahedron Lett. 1993, 34, 1303;
- (b) Chang, S. Y.; Jiaang, W. T.; Cherng, C. D.; Tang, K. H.; Huang, C. H.; Tsai, Y. M. J. Org. Chem 1997, 62, 9089;
- (c) Jiaang, W. T.; Lin, H. C.; Tang, K. H.; Chang, L. B.; Tsai, Y. M. J. Org. Chem 1999, 64, 618.
- 37. (a) Giese, B.; Angew. Chem. Int. Ed. Engl. 1985, 24, 553;
- (b) Curran, D. P.; Jiaang, W. T.; Palovich, M.; Tsai, Y. M. Synlett 1993, 403.
- 38. Tsai, Y. M.; Tang, K. H.; Jiaang, W. T. Tetrahedron Lett 1996, 37, 7767.
- 39. Siedem, C. S.; Molander, G. A. J. Org. Chem 1996, 61, 1140.
- 40. Saleur, D.; Bouillon, J. P.; Portella, C. Tetrahedron Lett 1999, 40, 1885.
- 41. Saleur, D.; Bouillon, J. P.; Portella, C. Tetrahedron Lett 2000, 41, 321.
- 42. Tsai, Y. M.; Cherng, C. D.; Nieh, H. C.; Sieh, J. A. Tetrahedron 1999, 55, 14587.
- 43. Danheiser, R. L.; Fink, D. M. Tetrahedron Lett. 1985, 26, 2513.
- 44. Takeda, K.; Nakajima, A.; Takeda, M.; Okamoto, Y.; Sato, T.; Yoshii, E.; Koizumi, T.; Shiro, M. J. Am. Chem. Soc. 1998, 120, 4947.
- 45. (a) Takeda, K.; Ohtani, Y. Org. Lett 1999, 1, 677.
- (b) Takeda, K.; Tanaka, T. Synlett 1999, 705.
- 46. Reich, H.; Holtan, R. C.; Bolm, C. J. Am. Chem. Soc. 1990, 112, 5609.
- 47. (a) Barbaro, G.; Battaglia, A.; Giorgianni, P.; Maccagnani, G.; Macciantelli, D.; Bonini, B. F.; Mazzanti, G.; Zani, P. J. Chem. Soc., Perkin Trans. 1 1986, 381.
- (b) Capperucci, A.; Degl'Innocenti, A.; Ricci, A.; Reginato, G. J. Org. Chem 1989, 54, 19.
- 48. Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Peri, F.; Ricci, A. J. Chem. Soc., Perkin Trans. 1 1996, 2803.
- 49. (a) Bonini, B. F.; Franchini, M. C.; Mazzanti, G.; Ricci, A.; Fauzza, L. R.; Zani, P. Tetrahedron Lett. 1994, 35, 9227.
- (b) Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A. Tetrahedron 1996, 52, 4803.
- 50. Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A. Tetrahedron 1997, 53, 7897.
- 51. Bonini, B. F.; Franchini, M. C.; Fochi, M.; Mazzanti, G.; Ricci, A.; Varchi, G. Tetrahedron Lett 1999, 40, 6473.
- 52. Brillon, D. Sulfur Rep. 1992, 12, 297.
- 53. (a) Royon, R. P.; Portella, C. Tetrahedron Lett 1996, 37, 6113.
- (b) Brigaud, T.; Lefebvre, O.; Royon, R. P.; Portella, C. Tetrahedron Lett 1996, 37, 6115.
- 54. Brigaud, T.; Doussot, P.; Portella, C. J. Chem. Soc. Chem. Commun 1994, 18, 2117.
- 55. Saleur, D.; Brigaud, T.; Bouillon, J. P.; Portella, C. Synlett 1999, 432.
- 56. Lefebvre, O.; Brigaud, T.; Portella, C. Tetrahedron 1998, 54, 5939.
- 57. Lefebvre, O.; Brigaud, T.; Portella, C. Tetrahedron 1999, 55, 7233.
- 58. Bonini, B. F.; Franchini, M. C.; Mazzanti, G.; Ricci, A.; Sala, M. J. Org. Chem. 1996, 61, 7242.
- 59. Kuwajima, I.; Matsumoto, K. Tetrahedron Lett 1979, 4095.
- 60. Horiuchi, Y.; Oshima, K.; Utimoto, K. J. Org. Chem 1996, 61, 4483.
- 61. Horiuchi, Y.; Taniguchi, M.; Oshima, K.; Utimoto, K. Tetrahedron Lett 1995, 36, 5353.
- 62. Mochida, K.; Okui, S.; Ichikawa, K.; Tsuchiya, T.; Yamamoto, K. Chem. Lett 1986, 805.
- 63. Miller, J. A.; Zweifel, G. J. Am. Chem. Soc 1981, 103, 6217.
- 64. Linderman, R. J.; Chen, K. Tetrahedron Lett 1992, 33, 6767.
- 65. Yohida, J.; Matsunagua, S.; Isoe, S. Tetrahedron Lett 1989, 30, 5293.
- 66. Salzmann, T. N.; Ratcliffe, R. W.; Christensen, B. G.; Bouffard, F. A. J. Am. Chem. Soc 1980, 102, 6161.
- 67. Sakaguchi, K.; Mano, H.; Ohfune, Y. Tetrahedron Lett 1998, 39, 4311.
- 68. Patrocínio, A. F.; Moran, P. J. S. Synth. Commun 2000, 30, 1419.
- 69. Brook, A. G.; Pierce, J. B. Abst. 149th Meeting Am. Chem. Soc. Detroit, 1965, p.2P.
- 70. Brook, A. G. Acc. Chem. Res 1973, 6, 77.
- 71. Trommer, M.; Sander, W. Organometallics 1996, 15, 189.
- 72. Brook, A. G.; Pierce, J. B.; Duff, J. M. Can. J. Chem 1975, 53, 2875.
- 73. (a) Soderquist, J. A.; Anderson, C. L.; Miranda, E. I.; Rivera, I.; Kabalka, G. W. Tetrahedron Lett 1990, 31, 4677.
- (b) Buynak, J. D.; Geng, B.; Uang, S.; Strickland, J. B. Tetrahedron Lett 1994, 35, 985.
- 74. Buynak, J. D.; Strikcland, J. B.; Lamb, G. W.; Khasnis, D.; Modi, S.; Williams, D.; Zhang, H. J. Org. Chem 1991, 56, 7076.
- 75. Takeda, K.; Ohnishi, Y.; Koizumi, T. Org. Lett 1999, 1, 237.
- 76. (a) Tacke, R.; Linoh, H.; Stumpf, B.; Abraham, W. R.; Kieslisch, K.; Ernest, L. Z. Naturforch 1983, 38b, 616.
- (b) Syldatk, C.; Stoffregen, A.; Wuttke, F.; Tacke, R. Biotech. Lett 1988, 10, 731.
- (c) Fischer, L.; Wagner, S. A.; Tacke, R. Appl. Microbiol. Biotechnol 1995, 42, 671.
- (d) Tacke, R.; Wagner, S.A.; Brakmann, S.; Wuttke, F. J. Organomet. Chem 1993, 458, 13.
- 77. Tacke, R.; Linoh, H. In The Chemistry of Organic Silicon Compounds; Patai, S.; Rappoport, Z., Eds.; John Wiley & Sons Ltd., New York 1989, chapter 18.
- 78. (a) Csuk, R.; Glanzer, B.I. Chem. Rev 1991, 91, 49.
- (b) Servis, S. Synthesis 1990, 1.
- 79. (a) Mitteilung, K.; Eichberger, G.; Faber, K.; Griengl, H. Monatsch. Chem 1985, 116, 1233.
- (b) Deardorff, D. R.; Myles, D. C.; Macferrin, K. D. Tetrahedron Lett 1985, 26, 5615.
- (c) Wendhausen, R.; Moran, P. J. S.; Joekes, I.; Rodrigues, J. A. R. J. Mol. Catal. B: Enzym. 1998, 5, 57.
- 80. Patrocínio, A. F.; Corręa Jr., I. R.; Moran, P. J. S. J. Chem. Soc. Perkin Trans. 1 1999, 3133.
- 81. Patrocínio, A. F.; Moran, P. J. S. J. Chem. Res. (S) 2000, 404.
- 82. (a) Bassindale, A. R.; Brook, A. G.; Jones, P. F.; Lennon, J. M. Can. J. Chem 1975, 53, 332.
- (b) Buynak, J. D.; Strikcland, J. B.; Hurd, T.; Pan, A. J. Chem. Soc. Chem. Commun 1989, 89.
- 83. Tamao, K.; Kakui, T.; Akita, M.; Iwahara, T.; Kanatani, R.; Yoshida, J.; Kumada, M. Tetrahedron 1983, 39, 983.
- 84. Brook, A. G. J. Am. Chem. Soc 1957, 79, 4373.
- 85. (a) Mori, A.; Fujita, A.; Ikegashira, K.; Nishihara, Y.; Hiyama, T. Synlett 1997, 693.
- (b) Wilson, S. R.; Hague, M. S.; Misra, R. N. J. Org. Chem 1982, 47, 747.
- (c) Barretm, A. G. M.; Hill, J. M.; Wallace, E. M.; Flygare, J. A. Synlett 1991, 764.
- (d) Hudrlik, P. F.; Abdallah, Y. M.; Kulkarni, A. K.; Hudrlik, A. M. J. Org. Chem 1992, 57, 6552.
- (e) Hiyama, T.; Obayashi, M. J. Org. Chem 1983, 48, 912.
- 86. (a) Linderman, R. J.; Ghannam, A. J. Am. Chem. Soc 1990, 112, 2392.
- (b) Linderman, R. J.; Chen, K. Tetrahedron Lett 1992, 33, 6767.
- 87. Danheiser, R. L.; Fink, D. M.; Okano, K.; Tsai, Y. M.; Szczepanski, S. W. J. Org. Chem 1985, 50, 5393.
- 88. Lipshutz, B. H.; Lindsley, C.; Susfalk, R.; Gross, T. Tetrahedron Lett 1994, 35, 8999.
- 89. Patrocínio, A. F.; Moran, P. J. S. Synth. Commun., in press.
- 90. Corey, E. J.; Seebach, D. Angew. Chem., Int. Ed. 1965, 4, 1075.
- 91. Brook, A. G.; Duff, J. M.; Jones, P. F.; Davis, N. R. J. Am. Chem. Soc. 1967, 89, 431.
- 92. Corey, E. J.; Seebach, D.; Freedman, R. J. Am. Chem. Soc 1967, 89, 434.
- 93. Gröbel, B. T.; Seebach, D. Synthesis 1977, 357.
- 94. Bouillon, J. P.; Portella, C. Tetrahedron Lett 1997, 38, 6595.
- 95. Suda, K.; Watanabe, J.; Takanami, T. Tetrahedron Lett. 1992, 33, 1355.
- 96. Patrocínio, A. F.; Moran, P. J. S. J. Orgamet. Chem 2000, 603, 220.
- 97. Katritzky, A. R.; Wang, Z.; Lang, H. Organometallics 1996, 15, 486.
- 98. Picard, J. P.; Calas, R.; Dunogučs, J.; Duffaut, N.; Gerval, J.; Lapouyade, P. J. Org. Chem. 1979, 44, 420.
- 99. Tongco, E. C.; Wang, Q.; Prakash, G. K. S. Synth. Commun. 1997, 27, 2117.
- 100. Fleming, I.; Roberts, R. S.; Smith, S. C. Tetrahedron Lett 1996, 37, 9395.
- 101. Still, W. C. J. Org. Chem 1976, 41, 3063.
- 102. Fleming, I.; Ghosh, U. J. Chem. Soc., Perkin Trans. 1 1994, 257.
- 103. Kuwajima, I.; Abe, T.; Minami, N. Chem. Lett 1976, 993.
- 104. Paredes, M. D.; Alonso, R. Tetrahedron Lett 1999, 40, 3973.
- 105. Nakada, M.; Nakamura, S.; Kobayashi, S.; Ohno, M. Tetrahedron Lett. 1991, 32, 4929.
- 106. Capperucci, A.; Degl'Innocenti, A.; Faggi, C.; Ricci, A. J. Org. Chem 1988, 53, 3612.
- 107. (a) Fleming, I.; Marchi Jr., D. Synthesis 1981, 560.
- (b) Ager, D.; Fleming, I.; Patel, S. K. J. Chem. Soc., Perkin Trans. 1 1981, 2521.
- 108. Bonini, B.F.; Franchini, M.C.; Mazzanti, G.; Passamonti, U.; Ricci, A.; Zani, P. Synthesis 1995, 92.
- 109. Yamamoto, K.; Suzuki, S.; Tsuji, J. Tetrahedron Lett 1980, 21, 1653.
- 110. Geng, F.; Maleczka Jr., R. E. Tetrahedron Lett 1999, 40, 3113.
- 111. Reich, H. J.; Kelly, M. S.; Olson, R. E.; Holtan, R. C. Tetrahedron 1983, 39, 949.
- 112. Brook, A. G.; Ionkin, A.; Lough, A. J. Organometallics 1996, 15, 1275.
- 113. Reich, H. J.; Kelly, M. J. J. Am. Chem. Soc. 1982, 104, 1119.
- 114. (a) Tius, M. A.; Hu, H. Tetrahedron Lett 1998, 39, 5937.
- (b) Stergiades, I. A.; Tius, M. A. J. Org. Chem 1999, 64, 7547.
- 115. Kuwajima, I.; Abe, T.; Minami, N. Chem. Lett 1976, 993.
- 116. Nowick, J. S.; Danheiser, R. L. Tetrahedron 1988, 44, 4113.
- 117. Shinokubo, H.; Oshima, K.; Utimoto, K. Tetrahedron 1996, 52, 14533.
- 118. Soderquist, J. A.; Hassner, A. J. Am. Chem. Soc. 1980, 102, 1577.
Publication Dates
-
Publication in this collection
19 Apr 2001 -
Date of issue
2001
History
-
Received
14 Nov 2000

