Abstracts
This paper intends to review the basic theory of Near Infrared (NIR) Spectroscopy and its applications in the field of Analytical Science. It is addressed to the reader who does not have a profound knowledge of vibrational spectroscopy but wants to be introduced to the analytical potentialities of this fascinating technique and, at same time, be conscious of its limitations. Essential theory background, an outline of modern instrument design, practical aspects, and applications in a number of different fields are presented. This work does not intend to supply an intensive bibliography but refers to the most recent, significant and representative material found in the technical literature. Because this paper has been produced as consequence of the First Workshop on Near Infrared Spectroscopy, whose venue was Campinas - Brazil, as a pre-conference activity of the XI National Meeting on Analytical Chemistry (ENQA), it also depicts the state of the art of NIR spectroscopy in Brazil, pointing out the current achievements and the need to take the technology to a level consistent with this country's economical activities.
near-infrared spectroscopy; chemometrics; instrumentation; analytical applications
Este trabalho visa prover uma revisão da teoria da Espectroscopia no Infravermelho Próximo (NIR) e das suas aplicações no campo das Ciências Analíticas. Ele é dirigido ao leitor que não possui um conhecimento profundo sobre espectroscopia vibracional mas que gostaria de ser introduzido às potencialidades analíticas desta técnica fascinante e, ao mesmo tempo, se tornar consciente das suas limitações. A teoria fundamental, uma revisão dos instrumentos modernos existentes e aplicações em inúmeros campos, são apresentados. Este trabalho não pretende suprir uma revisão bibliográfica intensiva mas se refere aos materiais recentes mais significativos e representativos encontrados na literatura técnica. Uma vez que este texto foi produzido em conseqüência do primeiro "workshop" em espectroscopia NIR, que ocorreu em Campinas - Brasil, como uma atividade preliminar ao 11º Encontro Nacional de Química Analítica (ENQA), ele também apresenta o estado da arte na Espectroscopia NIR no Brasil, mostrando seus resultados mais recentes e apontando a necessidade de se elevar esta tecnologia a um nível consistente com aquele de suas atividades econômicas.
REVIEW
Near Infrared Spectroscopy: fundamentals, practical aspects and analytical applications
Celio Pasquini
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13084-971 Campinas - SP, Brazil
Correspondence Correspondence to Celio Pasquini e-mail: pasquini@iqm.unicamp.br
ABSTRACT
This paper intends to review the basic theory of Near Infrared (NIR) Spectroscopy and its applications in the field of Analytical Science. It is addressed to the reader who does not have a profound knowledge of vibrational spectroscopy but wants to be introduced to the analytical potentialities of this fascinating technique and, at same time, be conscious of its limitations. Essential theory background, an outline of modern instrument design, practical aspects, and applications in a number of different fields are presented. This work does not intend to supply an intensive bibliography but refers to the most recent, significant and representative material found in the technical literature. Because this paper has been produced as consequence of the First Workshop on Near Infrared Spectroscopy, whose venue was Campinas Brazil, as a pre-conference activity of the XI National Meeting on Analytical Chemistry (ENQA), it also depicts the state of the art of NIR spectroscopy in Brazil, pointing out the current achievements and the need to take the technology to a level consistent with this country's economical activities.
Keywords: near-infrared spectroscopy, chemometrics, instrumentation, analytical applications
RESUMO
Este trabalho visa prover uma revisão da teoria da Espectroscopia no Infravermelho Próximo (NIR) e das suas aplicações no campo das Ciências Analíticas. Ele é dirigido ao leitor que não possui um conhecimento profundo sobre espectroscopia vibracional mas que gostaria de ser introduzido às potencialidades analíticas desta técnica fascinante e, ao mesmo tempo, se tornar consciente das suas limitações. A teoria fundamental, uma revisão dos instrumentos modernos existentes e aplicações em inúmeros campos, são apresentados. Este trabalho não pretende suprir uma revisão bibliográfica intensiva mas se refere aos materiais recentes mais significativos e representativos encontrados na literatura técnica. Uma vez que este texto foi produzido em conseqüência do primeiro "workshop" em espectroscopia NIR, que ocorreu em Campinas Brasil, como uma atividade preliminar ao 11º Encontro Nacional de Química Analítica (ENQA), ele também apresenta o estado da arte na Espectroscopia NIR no Brasil, mostrando seus resultados mais recentes e apontando a necessidade de se elevar esta tecnologia a um nível consistente com aquele de suas atividades econômicas.
1. Introduction and Historical Overview
Near Infrared Spectroscopy (NIR) is a type of vibrational spectroscopy that employs photon energy (hn) in the energy range of 2.65 x 10-19 to 7.96 x 10-20 J, which corresponds to the wavelength range of 750 to 2,500 nm (wavenumbers: 13,300 to 4,000 cm-1). This energy range is higher than necessary to promote molecules only to their lowest excited vibrational states (through a fundamental vibrational transition) and lower than typical values necessary for electron excitation in molecules (except for some rare earth compounds). Its overall objective is to probe a sample in order to acquire qualitative and/or quantitative information coming from the interaction of near-infrared electromagnetic waves with its constituents.
The analytical methods resulting from the use of the NIR spectroscopic region reflect its most significant characteristics, such as: fast (one minute or less per sample), non-destructive, non-invasive, with high penetration of the probing radiation beam, suitable for in-line use, nearly universal application (any molecule containing C-H, N-H, S-H or O-H bonds), with minimum sample preparation demands. The combination of these characteristics with instrumental control and data treatment has made it possible to coin the term Near-Infrared Technology.1
However, in order to have such a type of spectroscopy and to evolve it into a useful analytical technique, it was first necessary to discover the radiation in the NIR energy range, which is invisible to the naked eye. However, this did not constrain the clever mind of the German born, English scientist, Frederick William Herschel.
Sir Herschel was an astronomer and musician who, in between composing and discovering the planet Uranus, also marked his place in science by running an experiment to find out the contribution of each of the colours arriving from dispersed white sunlight in increasing the temperature of the substances exposed to them. The experiment is cited as an example of scientific insight because Herschel did not stop probing what happen with the temperature when he reached the end of the visible red colour region of the dispersed light. Contrary to common sense, he continued observing what happened with the temperature, placing the thermometer beyond that point. Surprisingly, he found the temperature still rises. Herschel used blackened bulb thermometers and glass prisms which are transparent to short wave NIR radiation and reported his achievement by referring to this region as "calorific rays" found beyond the red. This region was later named infrared, using the Greek prefix "infra" which means "below". The first non-visible part of the electromagnetic spectra had then been reported to exist by the year 1800.2,3
Although NIR radiation had been detected before that of the medium infrared, it was this latter spectral region that first gained wide acceptance after the work initiated in 1900 by Coblentz, who was the first researcher to obtain absorbance spectra of pure substances and verify their usefulness for the identification of organic functional groups.
While medium infrared spectroscopy gained wide acceptance and constantly received both theoretical and instrumental progress, NIR spectroscopy was neglected by spectroscopists who, for a long time, could not find any additional attractive information in that spectral region which was occupied by broad, superimposed and weak absorption bands. In fact, this early ostracism, despite its potentialities, of the NIR spectral region was clearly demonstrated in a paper by Wetzel whose suggestive title is "Near-Infraed Reflectance Analysis - Sleeper Among Spectroscopic Techniques",4 published in 1983.
Nevertheless, the decade of the eighties has marked the "boom" of the technique. From 1930 to 1980 the total number of papers published dealing with NIR was about 255, while the following decade saw a number greater than 1000.5 Papers were produced with titles more adequate to the new reality of the technique and its increasing acceptation such as "Near-Infrared Spectroscopy - The giant is running strongly", in 19946 and "The history of near infrared spectroscopy analysis: Past, present and future From sleeping technique to the morning star of spectroscopy" in 1998.7 The present number of publications on the NIR technique is certainly greater than 15 000, which attests to its wide acceptance and reveal it as a fecund field for research. Such productivity led to the appearance, in 1993, of the Journal of Near Infrared Spectroscopy, the first periodical entirely dedicated to this field.
Pioneering work on the analytical exploitation of the NIR spectral region had been developed since 1938, when the determination of water in gelatin by employing its stronger absorption in the NIR region was described.8 Barchewitz was the first to apply NIR spectroscopy to the determination of fuel.9 Many other contributions can be found in the early days of NIR spectroscopy,10-12 all of them trying to identify a single wavelength which could be employed for quantitative determination, expecting Beer´s law to be obeyed.
Although the paper by Kubelka and Munk13 on diffuse reflection had been published before any practical application of NIR had been proposed, the significance of this type of measurement technique passed unnoticed by the analytical spectroscopists for a long period. However, efforts towards the development of instrumental methods for application to agriculture, initiated by the Department of Agriculture of the USA, would end with the recognition of the diffuse reflectance measurement mode as the most suitable for the kind of sample in which they were interested. At this point the figure of Karl Norris emerges as the most prominent person in the history of NIR spectroscopy.
Karl Norris initiated his work with NIR by searching for new methods for the determination of moisture in agricultural products, first by extracting the water in methanol14 and soon afterward by suspending ground seeds in CCl4. The first results of such an approach were published in 196515 and republished in a special issue of the Journal of Near Infrared Spectroscopy honouring Karl Norris in 1996.16 This paper presented the "way of thinking" of that pioneer researcher and describes the adoption of a two wavelength model to correlate the spectral data to the analyte concentration and, at same time, abandons Beer´s law as a pre-requisite for quantitative analysis (because it did not work in the highly scattering medium in which the transmitance was measured). Simultaneously, it paved the way to a more ambitious achievement, that was the use of diffuse reflectance as a non-destructive measurement in the NIR region, which makes it possible to work with the sample directly, without any pre-treatment. Further work by Ben-Gera and Norris consolidates the approach of using NIR spectroscopy and originated one of the most cited paper in the field.17
After the door had been opened and the potentialities demonstrated by the pioneers, NIR spectroscopy encountered fast development impelled mainly by instrumental (spectrophotometer) improvements associated with spectral data acquisition and their treatment (micro-computer) and which, to a great extent depended on the new discipline of Chemometrics,18 which supplies the tools for gathering information and its wise use. A recent review, has been published emphasising one of the main qualities of NIR spectroscopy: its rapid-response as an analytical tool.19
In Brazil, the first contributions to the field of applied analytical NIR spectroscopy can be traced back to 1991.20,21 They were produced in a Brazilian research institution and report on the direct determination of a natural sweetener, stevioside, in the leaves of a native South America plant. Possibly, other pioneering uses of NIR in Brazil were carried out. However, these have not been reported in a traceable publication. The attribution of the year of 1991 for the first Brazilian contribution to NIR is in agreement with the contents of the preface of the first edition (1987) of the book by Williams and Norris on NIR technology where is it can be found that "... we have no information (about NIR research) from Latin America or from the continent of Africa beyond knowing that a number of instruments have been located there".1
Perhaps some practical results achieved by the analytical use of NIR spectroscopy should be used at this point, to convince the reader of its usefulness and attractive aspects and motivate him/her to continue to be in contact with the principles and practical aspects of this amazing technique. For an analytical chemist trained in classical methodology, such as the author and many of his contemporary colleagues, perhaps one of the most significant and motivating examples is the replacement of a wet chemical procedure (the Kjeldhal method) for determination of protein in commodities (wheat, corn and soybean, for example), by the direct NIR method. The Kjeldhal method, as every analytical chemist well knows, is a quite robust and accurate method for determination of protein in a multitude of samples, mainly commodities. The method is based on the digestion of the ground grain in concentrated sulphuric acid containing a catalyst such as mercury or selenium. After an hour or so the digested product is made alkaline and submitted to a distillation. The distilled ammonia is collected in standard boric acid solution and, finally, the excess acid is titrated with standard sodium hydroxide solution. This cumbersome and time consuming procedure has been replaced by the NIR spectroscopic method which can directly determine the protein content of the whole or ground grain, through diffuse reflectance, in less than one minute, generating virtually no hazardous residue. The countries employing this technology can obtain a better pricing for their commodities which are 100% certified for their protein content.
On the other hand, it is also wise, in order to be impartial, to alert the reader that, as marvellous it can appear at first glance, NIR technology is and will be always heavily dependent on the existence of good and acceptable reference methods as the Kjeldhal method. That is because at the learning stage (modelling stage) the direct method based on NIR needs to be able to identify the spectral characteristics or which combination of those characteristics are to be correlated for, in the above example, determining protein content in grain. The difference between failing or succeeding in this task is greatly dependent on the quality of the reference values associated with the samples in the training set. Nevertheless, once the learning stage is concluded, the final result is perhaps the closest that our present technology is able to produce of an ideal analytical method.
2. Theory
In order to access the origin of a NIR spectra, to be able to interpret it and have an important tool to guide in analytical method development, one should be familiar with the fundamentals of vibrational spectroscopy. The NIR spectrum originates from radiation energy transferred to mechanical energy associated with the motion of atoms held together by chemical bonds in a molecule. Although many would approach method development in a purely empirical way, knowledge of the theory can help to look at the important wavelengths and quicker optimisation of the modelling stage.
2.1. Vibrational spectroscopy
At ambient temperature most of the molecules are in their fundamental vibrational energy levels. Atoms or group of atoms participating in chemical bonds are displacing one in relation to the other in a frequency that is defined by the strength of the bond and the mass of the individual bonded atoms or their groups. The amplitudes of these vibrations are of a few nanometers and will increase if some energy is transferred to the molecule. This energy can be transferred from a photon of a given wavelength (l), for which the energy (Ep) can be given by:

in which h is the Planck constant and c is the velocity of light.
2.2. The classical mechanical model for a diatomic molecule
The simplest classical model employed to have a didactic insight on the interaction of radiation and matter in the NIR spectral region depicts a diatomic molecule as two spherical masses (m1 and m2) connected with a spring with a given force constant (k). Hook´s law states that the energy (E) of this system is given by:

where m is the reduced mass:

The molecular vibration can be described by a simplified model supposing a harmonic oscillator for which the potential energy (V), as a function of the displacement of the atoms (x), is given by:

Figure 1A shows the behaviour of the potential energy as a function of atom displacement from the equilibrium (minimum energy) position. This first approach is useful to understand the concept of vibrational energy. However, it fails when a microscopic system such as molecules is being considered. The failure arises from the fact that molecular systems can not assume the continuous energy profile predicted by the classical "balls-on-spring" model. The molecular system can only have some discrete energy levels (Eu) defined by quantum mechanics by the equation:

where u is the vibrational quantum number, Eu is the energy associated with the uth quantum level and n is the fundamental vibrational frequency. In the classical model this frequency is defined by:

An additional restriction imposed by the harmonic/quantum model is that the transitions can only occur between adjacent levels, Du = ± 1. Furthermore, for this model, the difference of energy between two adjacent states is always the same (see Fig. 1A).
The energy of the electromagnetic radiation that is absorbed in order to promote the molecule to an excited level should match the difference between two adjacent energetic levels. Therefore, the photon energy must be

For the harmonic oscillator Du = 1 and Ep = hn, which matches the predicted equal energy difference between one state and the other of immediately higher energy. Figure 2 shows the effect of photon absorption on the energy and amplitude of vibration.
The classical analogue to this behaviour is the concept of resonance. In this concept, the physical characteristics of a "string" stretched between two supporting points, such as its linear density and the force by which it is stretched, will define its natural frequency of vibration (as a guitar string does). The amplitude of this natural vibration (therefore, its energy) can be increased by exposing the string to an acoustic wave propagating in the air, with the same frequency, produced, for example, by a distant stroked string with the same characteristics. The first string undergoes no energy change if the acoustic wave frequencies and the natural frequency do not match each other. Similarly, only radiation of a certain frequency (and wavelength) can excite the vibrational levels of molecules. However, this model fails in the molecular world because it is not a quantum model. In the "string world" , the energy they can obtain from the exciting mechanical wave can increase continuously while a "quantum string" is able to vibrate at only a given frequency and at only some pre-defined amplitude.
Although the harmonic model can help understanding vibrational spectroscopy, it produces some disappointing restrictions for NIR spectroscopy because it can not permit transitions where Du is greater than 1. Transitions with Du = 2 or greater are forbidden by the harmonic/quantum model and, in this way, most of the observable phenomena in the NIR region, the overtones bands, should not exist. Also, the vibrations in the harmonic model are independent and their combinations would not exist under the restrictions imposed by the model. Nevertheless, both overtones and combination bands exist.
2.3. The anharmonic model
Figure 1B shows a more realistic mechanical model for a diatomic molecule. The molecule is still approximated by two balls connected with a spring. However, the model considers some non-ideal behaviours of the oscillator which account for repulsion between electronic clouds when the atomic nuclei approach (notice how the potential energy rises fasten than in the harmonic model) and a variable behaviour of the bond force when the atoms move apart from one another. In fact, in a real molecule, the over displacement ("strengthening of the spring") of the atomic nuclei will cause molecule bond rupture with consequent dissociation of the atoms. A complex function of the potential energy is assumed to describe the last effect which can be approximated by using higher order terms of displacement, as depicted in the equation

A function that approximates the anharmonic behaviour of a diatomic molecule is the Morse function that describes the potential energy of the molecule using the equation:

where a is a constant for a given molecule, De is the spectral dissociation energy, re is the equilibrium distance between the atoms and r is the distance between the atoms at any instant.
Applying quantum mechanics to the Morse equation results in the vibrational levels being described by the equation:

in which xm is the anharmonicity constant of the vibration, whose value is between 0.005 and 0.05.
The anharmonic/quantum model also imposes some restrictions on the possible energy states of the molecules. However, it predicts the occurrence of transitions with Du = 2 or higher and the existence of combination bands between vibrations. These two types of bands are the most common absorption bands in the NIR spectral region. It also predicts that the separation between two adjacent energy levels decreases with u, the vibrational quantum number.
Under the assumptions of the anharmonic model, the vibrations are no longer independent of each other and can interact with one another. Therefore, the total vibrational energy (Eu) contains cross-terms from more than one vibration in the molecule:

for r £ s; in which nr and ur are the fundamental frequency and the quantum number of vibrational mode r, respectively, and xrs is the anharmonicity constant for the interaction of vibrational modes r and s.
Anharmonicity can also be present in the electrical properties of a molecule. Specifically, it affects its dipole moment which, in an anharmonic model, does not have a linear dependence with the interatomic distance. This kind of anharmonicity can provide the way for overtones and combination bands to occur even if no mechanical deviation of the harmonic model is observed for a given system.
2.4. Origin and intensity of a NIR absorption band
So far, it is possible to understand from theory that radiation of a given frequency, capable to supply exactly the energy between two vibrational levels or of their overtones or combinations of two or more vibrations, can be absorbed by the molecule and can produce excitation to a higher vibrational energy level. The match of radiation energy with the energy difference between two vibrational levels causes a selective response of the molecular system to the incident radiation. It means that in a given wavelength range, some frequencies will be absorbed, others (that do not match any of the energy differences possible for that molecule) will not be absorbed while some will be partially absorbed. This complex figure of the intensity of absorption versus wavelength constitutes the absorption spectra of a substance or sample.
However, only the energy match between photons and vibrational levels is not sufficient for radiation absorption. For a vibration to be active, it is necessary that the electrical oscillating field of the electromagnetic wave (light) can interact with the molecule. This can only occur if the displacement of the atoms in a vibrational mode can produce a change in the dipole moment of the molecule or in the local group of vibrating atoms. While this is true considering a fundamental mode, it is worth noting that, for combination bands permitted by anharmonicity, it would be necessary that only one of the combining vibrations be active (causing dipole change). This feature may cause some vibrations, which can not be observed in the middle infrared, to be displayed by a NIR spectrum.
The intensity of a given absorption band is associated with the magnitude of the dipole change during the displacement of atoms in a vibration and with its degree of anharmonicity. Both phenomena are present in great intensity associated with bonds involving the hydrogen atom and some other heavier element such as carbon, nitrogen and sulphur. The O-H, C-H, N-H and S-H bonds tend to present high anharmonicity and high bond energy with fundamental vibrational transitions in the region of 3000 4000 nm. Therefore, it allows to predict the overtones and combinations of the fundamental vibrations of such bonds to occur in the region of energy associated with NIR photons. Intensities are in between 10, for combinations, up to 1000, for successive overtones, times lower than the absorption resulting from fundamental vibrations.
The spectral occurrences in the NIR region are dominated by overtones and combination absorption bands. However, some other effects, associated with the higher order terms in equation 8, may be observed and contribute to the complexity of the NIR spectrum. Coupling or resonance between different vibrations of the same functional group can occur as a function of the third and fourth order terms of equation 8.
Fermi resonance occurs between a fundamental and an overtone when their difference in energy is very low. This type of resonance results in a greater separation between the position of the two bands and in the intensification of the overtone band. Darling-Dennison resonance may promote the interaction between two high level overtones of a molecule and a combination band, for example, and is particularly intense for bonds containing hydrogen atoms. The complexity of the combination spectral region in the NIR spectrum of hydrocarbons is partly due the possibility of resonance between the combination bands and high order overtone for C-H bonds.
The primary practical consequence of both types of resonance on a NIR spectrum is the possibility of the appearance of two instead of one band in the combination region (1600 2500 nm). Additional information on the theory regarding vibrational NIR spectroscopy is found in more detail in several references.1, 5, 22 24
3. Instrumentation
A NIR spectrophotometer can be assembled with optical components employed for UV-Visible instruments. This fact imparts a lower cost to the NIR instrument, when compared with the mid-infrared (MIR) spectrophotometer. When necessary, as when a long optical guide needs to be employed to probe a distant sample, low OH containing materials should be employed. Furthermore, the instrument is, overall, more robust because its optical parts are not harmed by environmental humidity. MIR instruments, at present, have similar characteristics but they are far more expensive.
The most frequently employed detectors for the NIR spectral region are based on silicon, PbS and InGaAs photoconductive materials. In particular, the latter possess a very high detectivity (D*) and a very high response speed. Together with high powered radiation sources (a tungsten coil or a halogen lamp is employed by the majority of manufacturers) these detectors can impart a very high signal-to-noise ratio for NIR measurements. This fact partially compensates for the lower intensities of NIR absorption bands.
Table 1 shows a classification of modern NIR instruments in terms of the technology employed for wavelength selection. Many of the instruments are available commercially, although a few only exist on the researchers' benches.
3.1. Filter based instruments
Instruments employing filters as wavelength selectors are commercially available for dedicated applications. For example, an instrument for determination of the quality parameters of gasoline (Zeltex Inc.) employs 14 interference (Fabri-Perrot) filters and 14 LED (Light Emitting Diode) sources in the NIR region. Filter based instruments, although commercially available, are not described in detail in the literature, with the exception of early instruments.1
A two25 and three26 filter-based instruments dedicated to the identification of polymers for recycling purposes and for the determination of proteins and nitrogen, respectively, have been described recently. Such examples demonstrate the capabilities of dedicated instruments, which have found their place when a high demand application of NIR spectroscopy is identified.
Variable (scanning) interference filters based on the variation of the dielectric layer thickness by using piezoelectric drivers have recently been described.27 Although no device has been described to work in the NIR spectral region, it is possible that in the near future these devices will also operate in this region, becoming another alternative for robust, portable and rapid scan instruments.
3.2. LED based instruments
Recently a tendency to reduce the price and size of the instruments, aiming for use in the field, has taken advantage of the use of Light Emitting Diodes (LED). These devices can produce NIR radiation with a band width of about 30 - 50 nm, centred in any wavelength of the spectral region. The instruments can employ a set of LEDs as sources of narrow bands of near infrared radiation28-30 or use them to produce a polychromatic, highly stable source whose radiation is dispersed by using common monochromator devices such as those based on gratings or filter optics.31,32
Common LEDs can be purchased at prices lower than US$ 10 for the shorter NIR wavelength range (700 1100), although the LEDs operating at higher wavelengths are still expensive. LED and filter based instruments are in strict agreement with the modern tendency for low cost, specific applications and portable instrumentation for in field analysis.
3.3. AOTF based instruments
The instruments based on Acousto-Optical Tunable Filters (AOTF)33 are modern scan spectrophotometers employing a technology that allows constructing instruments with no moving parts, capable of reaching very high scan speeds over a broad range of the NIR spectral region. If necessary, random access to any number of wavelengths necessary to perform a given analytical determination can be easily implemented. Scan speed is fast and up to 2000 wavelengths can be selected per second. The scan speed is usually limited by the detector response time.
The AOTF operating in a non-collinear configuration (acoustic wave and radiation beam propagating at almost perpendicular angles), shown schematically in Figure 3, is a device made of a birefringent crystal of TeO2, cut in a special angle.34 Other materials can be used but the characteristics of TeO2 are suitable for the NIR regions and has been chosen by all instrument manufacturers. A piezoelectric material (usually LiNiO4) is attached to one end of the crystal which, under excitation from an external radio frequency signal (rf), produces a mechanical (acoustic) wave which propagates through the crystal. The acoustic wave produces a periodic variation of the refractive index of the crystal in a frequency determined by the rf signal, in the range of 50 to 120 MHz. The interaction of the electromagnetic wave and the acoustic wave causes the crystal to refract selectively a narrow wavelength band. The relationship between the diffracted radiation wavelength (l) and the frequency of the acoustic wave (fa) is given by:

where Dn is the difference in the refractive indexes of the birefringent TiO2 crystal, ua, is the velocity of the acoustic wave and a is a parameter dependent on the AOTF design.
The birefringence of the TeO2 crystal leads to the production of two monochromatic beams whose angular separation is about 7o . Both or only one diffracted beam can be used by NIR instruments. Dual beam (with a reference beam produced by splitting one of the beams) or single beam instruments can be found. Alternatively, the two monochromatic beams can be employed for the construction of a dual beam instrument with an optimised use of the radiation throughput.35
The non-moving parts concept of the AOTF-based NIR spectrophotometers impart to them some unbeatable qualifying characteristics for use in the field or on the factory floor, aiming at in-line monitoring. The wavelength precision is about ±0.05 nm and the resolution is dependent on the wavelength, with typical values in the range 5 to 15 nm for the wavelength in the range 1000 to 2500 nm.
3.4. Dispersive optics-based instruments
Dispersive instruments based on diffraction gratings were employed in the early days of NIR spectroscopy and were responsible for the research initially developed to consolidate NIR spectroscopy as an analytical tool. This technology has continued through the 90's and is still being used, with many manufacturers offering instruments with performance suitable for many practical applications.36 The first commercial Brazilian-made NIR spectrophotometer, manufactured by Femto Ltda., was launched in 1999 and is based on a monochromator employing a moving diffraction grating and Czerny-Turner optics. Its performance suffices for many transmitance measurement-based analytical methods employed in the control of sugar and alcohol production, a very important Brazilian industrial sector.
The instruments based on grating monochromators present the advantage of a relatively low cost when compared with other scanning instruments employing modern technologies. The main disadvantages of dispersive instruments are the slow scan speed and a lack of wavelength precision, which deteriorates for long term operation due to mechanically driven mechanism fatigue. This can be a negative regarding maintenance of multivariate models. Also, the presence of moving parts limits the use of dispersive instruments in the field and in more aggressive environments.
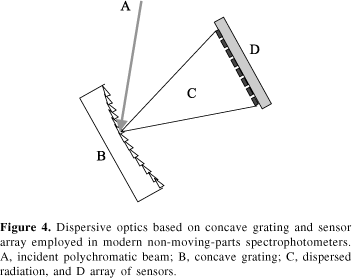
On the other hand, recent evolution in sensor production technology gives dispersive optics a longer life. That is because it is now possible to construct linear arrays of PbS and InGaAs sensors containing up to 256 independent elements. Placed in the focal plane of plane or concave grating optics, the sensor array allows to scan an entire spectra in a few milliseconds. Furthermore, as can be observed in Figure 4, no moving parts are present and the instrument can be placed, for example, on a truck to monitor the content of protein, oil and humidity in real time during grain harvest.37
The dispersive optics can also be improved regarding the signal-to-noise ratio, by making use of a Hadamard multiplexed design spectrophotometer.38,39 This type of instrument is not commercially available but it can impart a higher scan speed (about 10 seconds scan-1) and simplify the mechanics necessary for spectrum acquisition. The theoretical gain, Gsnr, in the signal-to-noise ratio of an individual signal after Hadamard transformation, for detector-limited noise, is given by:

where N is number of elements (wavelengths) multiplexed by the Hadamard optics.40 A dual beam Hadamard instrument constructed to operate in the NIR spectral region has been recently described.41 Despite its advantages, the Hadamard NIR spectrophotometer is still awaiting commercial interest in order to move its research achievements to industrial design and to become a technology accessible for end users.
Recently the Hadamard multilexing approach has been combined with modern AOTF technology. This junction was possible because modern radio frequency synthetizers can mix a number of signals of distinct frequencies and apply the resulting acoustic wave to the AOTF. Therefore, an optical signal composed of seven different wavelengths will result. In the original work, the authors describe a system where several radio frequency signals can be mixed according the Hadamard simplex matrix.42 A gain in the signal-to-noise ratio, as expected by Hadamard multiplexing, and an improved resolution resulting from the lower power per wavelength applied to the AOTF were observed.43
3.5. Fourier-transform based instruments
Spectrophotometers based on the use of interferometers and Fourier transform to recover the intensities of individual wavelengths in the NIR region are, undoubtedly, the instruments combining most of the best characteristics in terms of wavelength precision and accuracy, high signal-to-noise ratio and scan speed (although slower than AOTF based instruments).44 These instruments add to the previously mentioned Fellget gain the Jaquinot gain, which is not attainable with dispersive Hadamard instruments. The Jaquinot gain arises from the high radiation throughput achieved for a Fourier instrument, due the fact that it does not employ entrance or exit slits to limit the radiation intensity reaching the detector. Typical wavelength accuracy is better than 0.05 nm and the resolution can achieve values below 1 nm in the NIR region, at cost of decreasing the scan speed.
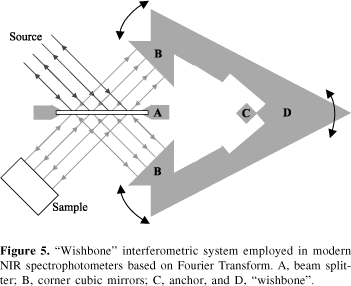
These advantages turn the NIR spectrophotometer based on interferometric measurement and Fourier transform into an unbeatable research instrument. On the other hand, the spectrophotometer is not as robust as an AOTF-based instrument which is assembled without any moving parts. The robustness of the Fourier spectrophotometer has been improved by using a "wishbone" type of interferometer, such as the one found in the Bomem instruments and shown in Figure 5. This type of interferometer employs half cubic mirrors to compensate for beam travelling distance changes arising from non uniform mirror movement during data acquisition. The result is a Fourier-based spectrophotometer that can tolerate some environmental vibrations without losing wavelength precision and photometric reproducibility. The price of a Fourier based instrument is comparable with the AOTF-based spectrophotometer and, therefore, both are considered expensive relative to other options.
3.6. Instrument selection criteria
Instrument selection must be guided by end application. Low cost instruments, based on filters and LEDs, suffice for many dedicated laboratory and routine in field applications. Instruments based on fixed dispersive optics and sensor arrays have proven to be a robust solution when multi-wavelength spectral data for in field applications are required.
Fourier-based and AOTF-based instruments must be the choice when research, wide application spectra and calibration transference are of concern. The AOTF spectrophotometer presents the most rapid random accesses to various wavelengths, which can be useful for certain applications for in-line monitoring, while the Fourier-based instruments show, on average, the best resolution and signal-to-noise ratios.
The modern NIR spectrophotometer requires a microcomputer in order to control the data acquisition system. The same tool is required for multivariate data treatment aiming at the development of new analytical methods. Also, for large projects involving more than one plant of an industrial company, the criterion must include the capability of data communication and transference through long distances, in order to exchange methods and other information among the various centers.
For use in research, where some unusual characteristics frequently become important, one should be aware of the facilities supplied by the instrument manufacturer allowing to access and control the instrument through clearly described routines and how to include such routines into a user-developed software. Standard digital data communication ports, such as USB, RS-232, RS-488 and IEEE-488, help to achieve instrument control if the operating manuals contain detailed information on the data exchanging protocols between the computer and the instrument. In terms of data treatment it is very important both to access the format in which the spectral data are saved (usually a manufacturer's format) and to have the capability to save spectral data in many different formats accessible to other commercial chemometric software packages.
Versatility in terms of sampling accessories can be useful for an instrument dedicated to research. Reflectance, transflectance and the usual transmitance measurement modes must be provided by the instrument after minimal and easy changes in its configuration. The use of fiber optic probes is desired, as many modern applications are based on their intensive use, in order to facilitate data acquisition routines.
Fiber optics can be embodied in an on-line monitoring project for any brand of instrument. The capacity of multiplexing a single instrument for monitoring many points in a process in a plant up to 100 m distant from the instrument is welcome. However, proof of long term stability of the multiplexing system must be given before a decision is taken and a multivariate model constructed.
The user must trade the benefits of the instrument's characteristics with the lowest cost. In time, for industrial users, the technical support given by the instrument supplier may make the difference to developing or not a successful NIR application. Previous history in NIR technology and the experience of the manufacturer in the field of interest are good starting points to consider when buying an instrument.
4. Practical Aspects of NIR Technology
A successful application of NIR technology in the analytical field depends on a series of equally relevant factors. Most of the advantages of NIR spectroscopy come from the possibility of using intact samples presented directly to the instrument without any pre-treatment. This fact implies in promoting non-conventional interaction of the radiation with matter in order to extract the spectral information and generate many different measurement modes.
4.1. Measurement modes
NIR spectrometry started as an unique technique when Karl Norris proposed that the spectral measurement could be obtained by analysing the information content of that portion of radiation diffusely reflected by solid samples instead of the weaker signal of transmitance. Today, diffuse reflectance is one of the various possibilities for employing the NIR spectral region. Figure 6 depicts the most common measurement modes employed by NIR spectroscopy.
Transmitance (Figure 6a) is obtained as in conventional UV-VIS spectroscopy. Transparent samples are measured in glass/quartz cuvetes with typical optical paths varying from 1 to 50 mm. The optical path is, in principle, defined by the spectral region being probed. It goes from a larger to a shorter path as the wavelength goes from the higher order overtones to the combination region around 2200 nm. Figure 6b shows a special way to obtain a transmitance measurement which is referred to as transflectance. This measurement mode is frequent when optical bundle probes are employed. The difference in relation to a simple transmitance measurement is in doubling the optical path as the radiation beam passes twice through the sample.
Many substances in solution follow Beer´s law, showing a linear relationship between concentration and absorbance. The best reference substance for transmitance and transflectance measurements should be carbon tetrachloride (CCl4), which shows no absorption bands in the whole NIR region. However, this toxic substance must be avoided unless it can be safely conditioned in a sealed flask. Empty cells and an internal reference beam have also been employed to calculate the transmitance (and absorbance) of transparent samples. Usually, these latter references cause a constant displacement of the baseline in relation to an ideal baseline at zero absorbance. The displacement is conveniently corrected if the first derivative of the spectra is used. It is important to be aware that the same limiting factors found in other spectral regions restrict the real systems from following Beer´s law over a wide concentration range. These limiting factors originate from the instrument and/or are characteristics of the sample constituents as, for example, the non-linearity of the detection system and the changes in hydrogen bond patterns as the concentration of the various species undergo relative concentration changes.
Diffuse reflectance measurement of solid samples (Figure 6c), as mentioned above, is a distinguishing measurement mode employed in NIR spectroscopy. In diffuse reflectance, scattering and absorbance by solid granules contribute to change the signal intensity. A rigorous treatment of the signal obtained in this type of measurement was established by Kubelka and Munk.13 The mathematical treatment results in the following equation that should replace Beer´s law, which is valid only for transparent homogeneous materials, and establishes a linear relationship between the concentration (C) and the diffuse reflectance (R):

where R is the reflectance which is obtained by:

where IR is the intensity of radiation reflected by the sample and IR0 the same quantity reflected by an non-absorbing material over the whole spectral range of measurement. The measurement of IR and IR0 requires the collection of the scattered radiation by an integration sphere, a perfectly reflecting reference (100% in all wavelengths) and, therefore, is seldom employed. In fact, the Kubelka-Munk equation is rarely employed, being substituted by more practical, although certainly non-linear, equations such as:

Equation 16 is much used for developing analytical methods based on reflectance measurements. The relationship does not depart much from the Kubelka-Munk prediction and, for small changes in the reflectance (R) (which are common for many applications) can be assumed to present a linear behaviour with the concentration of the analyte.
Figure 6 shows two more measurement modes employed in NIR spectroscopy. In the interactance mode (Figure 6d) a higher probability is given to the incident beam to interact with the sample. Consequently, the emerging beam (collected at a place somewhat distant from the location of incidence) contains more information on the sample constituents and reflects better the actual composition of the sample. Figure 6e shows the transmitance measurement of dense solid samples. Recently, this type of measurement has been found to be appropriate for quantitative determination of the active principle of pharmaceutical tablets because the longer optical path, resulting from internal scattering, can provide information which is better correlated with the average sample content than the surface dominated diffuse reflectance signal. This optical path has been reported to be about 65 times greater than the thickness of the tablet.45 Stray light is of major concern when using transmitance through solid samples because a very small fraction of the incident radiation reaches the detector. Noticeable non-correlated artefacts would be obtained if the sample holder and detector housing are not arranged properly to avoid this problem. Samplers to perform transmitance measurement in solids can be purchased from the major instrument manufacturers. A series of sample presentation devices has recently been reviewed.46
4.2. Some NIR spectra
Figure 7 shows an absorption spectrum of chloroform (CHCl3), a very simple molecule presenting NIR spectroscopic features that can be attributed only to single C-H vibration modes, their combinations and overtones. The spectrum has been obtained in the author's laboratory using a FT instrument and is a result of averaging 100 scans with resolution of 4 cm-1. The nominal optical path was 1 mm. The reference was CCl4. Absorption bands due to combination and to first and second overtones of C-H bonding can be easily observed. The apparent simplicity of the spectra is dismissed by a closer look at the insert in Figure 7. This is the spectrum of CHCl3 obtained in the short wave region (850 1150 nm) after changing the optical path to a nominal value of 5 cm. Five complete absorption bands can now be clearly identified in a wavelength region where the first spectrum has, in principle, non-observable spectral features. In fact, the number of combination and overtone bands (of the 6 existing fundamental bands) of chloroform has been reported to be equal to 34, all with energies in the NIR region.47
Chloroform can be used as an example to illustrate the complicating effect of the NIR region on the spectrum. However, it is worth to mention that this same complicating effect that results in a spectrum of difficult interpretation adds information about that specific type of C-H bond.
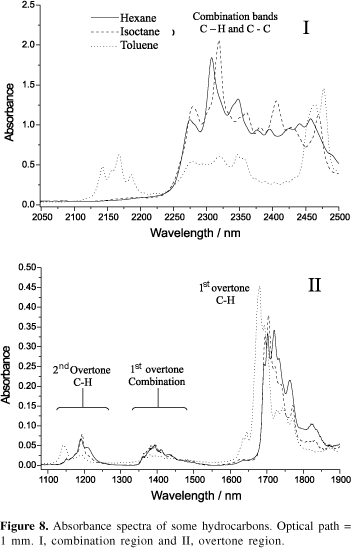
Figure 8 shows three hydrocarbon spectra, one of an aliphatic compound (hexane), one for a branched chain compound (isooctane) and one for an aromatic compound (toluene), obtained in the author's laboratory using a FT instrument with resolution of 4 cm-1, 1 mm optical path cell and in the range 1100 2500 nm. This set of spectra is of interest for applying NIR spectroscopy in fuel and petrol products analysis. The spectrum complexity, as could be anticipated, increases enormously and many superimposed bands can be observed. However, information on the different types of hydrocarbon are present throughout the NIR region. The combination, first and second overtones and overtones of combination regions of C-H bonds can be observed and the relative intensities can be compared, since the optical path has been held constant.
Figure 9 shows some diffuse reflectance spectra of representative materials. The spectra have been obtained in the author's laboratory using an AOTF-based instrument operating without optical fiber probes. In this instrument monochromatic radiation, isolated by the AOTF, is impinged directly onto the sample surface while the diffusely reflected radiation is collected by the InGaAs detector. Each spectrum is an average of 100 scans obtained in wavelength steps of 2 nm. Total acquisition time was 15 s. The reference was a disc of Spectralon®, a fluoro-polymer showing 100% reflectance in the NIR spectral region.
The spectra of an Aspirin tablet, virgin PET pellets, sugar from sugar cane and wheat flour (before and after partial drying), can be compared to identify their differences. The sugar spectra illustrates the effect of grain size as one of the factors affecting the reflectance spectrum. The log (1/R) signal has its intensity reduced as a consequence of the increasing specular fraction, which contains no information about the composition of the sample, as the sample grain size is reduced. On the other hand, reducing the grains of the sample to a more uniform size can help to distinguish the portion of the signal coming from the analyte itself from that caused by non-informative scattering.
The region containing the usual N-H absorption band at 2180 nm, employed for protein determination, can be visualised in the wheat spectrum.
4.3. Analytical information from NIR spectral data
Despite its complexity and the presence of broad and superimposed absorption bands, a quick look in the few NIR spectra shown in Figures 7, 8 and 9 reveals they have high information contents. This information content has been intensively exploited for qualitative and quantitative chemical and physical analytical purposes.
Sometimes the interest is in the use of the NIR spectrum as a source of information for qualitative identification of samples. The complexity of the spectrum forbids its direct use, as is done with the middle infrared region, where clear absorption bands can be easily distinguished and attributed, for example, to the presence of certain functional groups. Nevertheless, correlation charts showing where the absorption bands of O-H, C-H, N-H and S-H bonds of distinct compounds are located in the NIR spectral region can be found and used as a first approach for qualitative analysis.1,23 NIR spectroscopy is not suitable for structure elucidation. However, it has been widely employed for fast and direct access to identify starting products used, for example, by the pharmaceutical industry.
Interpretative NIR spectroscopy, or the use of such band characteristics as intensity and wavelength position for attribution of the origin of the spectral features, has been recommended as a support to guide and understand the results obtained for modern chemometric mathematical and statistical classification methods.23 In fact, the development of any analytical method based on NIR should be preceded by an exhaustive exercise aimed at correlating chemical knowledge about the sample, usually available beforehand, and spectroscopic features or vice-versa. The results of this exercise could anticipate spectral occurrences, their intensities, and the possible effect of parameters, such as temperature, on the NIR spectrum and the best wavelength regions to look for qualitatively and/or quantitatively relevant information. In the matter of interpretation of NIR spectra, two-dimensional correlation spectral analysis has emerged as a promising technique for helping complex spectral evaluation and band attribution.48,49
Table 2 summarises the sample properties, about which a NIR spectrum, in principle, is capable to give information, and some application examples. The information present in a NIR spectrum can be employed in the usual way to estimate the concentration of a given substance in a sample or to estimate a bulk or physical property when these can be, in any instance, reflected in significant changes in the intensity and/or wavelength of the spectral features produced by the sample. These features may be indirectly associated with the analyte, such as in the case, for example, of the determination of sodium chloride in concentrated aqueous solutions where the shift and intensity change of water absorption bands are the source of analytical information.50
5. Chemometrics
Since the early days, the use of NIR infrared spectral information for analytical purposes has relied on the multivariate approach for calibration. One of the very first papers employed the difference of absorbances at two wavelengths to correlate with the moisture content of grains.16 Nowadays this interdependence is of even more importance because of the increasing power of microcomputers and of the quality of the NIR spectrophotometers that allow thousands of data points to be obtained in a matter of seconds. Because NIR technology proposes a direct measurement of the sample without any pre-treatment, it also does not pose any inhibitory factor to data acquisition, contributing to the generation of large data sets during the calibration stage.
It is also clear, from the examples given above, that the complex nature of the NIR spectral region seldom permits the use of single wavelength models for quantitative purposes. In fact, only a few examples, for very simple binary systems such as water-ethanol, can be found in the literature.51 In the majority of cases the information necessary for a given analytical procedure needs to be gathered from among many variables, often reaching a number between 20 and 1000.
Currently, NIR spectroscopy is the analytical technique which most applies chemometrics. Chemometrics is the use of mathematical and statistical techniques for extracting relevant information from analytical data, in the present case, the NIR spectral data. Both Chemometrics and NIR technology have evolved in a symbiosis where NIR spectroscopy achieves more robust identification and quantitation models and extends its applicability, while posing new challenges to chemometrics that motivate the improvement of many of its techniques. An extensive review of the subject is out of the scope of this article. The reader is referred to reviews and textbooks which have treated this subject, some of them in direct connection with NIR spectroscopy.52-56
Meanwhile, it is relevant to know that a NIR spectral data set normally undergoes some type of pre-treatment before being used for qualitative or quantitative purposes. These data pre-treatments are used mainly to overcome problems associated with radiation scattering by a solid sample which is measured by reflectance and other spectrum base-line- affecting phenomena.
First and second derivatives of the original spectra have been used throughout57-59 and specifically designed data treatments, such as the Multiplicative Scatter Correction (MSC)60 and Standard Normal Variate (SNV)61 have shown success in dealing with the problem of scattered radiation.
An area that has undergone a notable evolution, regarding pre-treatment of NIR data, aims at the selection of relevant variables from a large multivariate set. Among others, it is possible distinguish the forward selection, backward elimination, stepwise procedures,54 Genetic Algorithm,62,63 and "jack knife" techniques.64 The use of variable selection procedures before the modelling step have demonstrated their value, contributing to achieving good models, better and more interpretable than is possible by indiscriminate use of all the variables.
Qualitative use of the NIR spectral information has become common, for example, for quality control of pharmaceuticals.65,66 Libraries containing NIR spectra of compounds of interest can be employed for positive identification with the aid of chemometric algorithms.56,67,68 The potentiality of NIR spectroscopy as an identity checking technique has been recognised in some pharmacopoeias.69,70
The most employed technique for qualitative analysis using NIR spectroscopy, supplied by many software packages, is based on Principal Component Analysis and is known as SIMCA (Software Independent Modelling Class Analogy).54
There is an arsenal of chemometric tools dedicated to make use of NIR spectroscopic information.53,54 The most common are Multiple Linear Regression (MLR), Principal Component Regression (PCR) and Partial Least Square Regression (PLS). All presuppose a linear relationship between the spectral data and the concentration or other property value to be determined.
PCR and PLS can be considered standard calibration techniques for NIR spectroscopy. The main advantage of these techniques is to avoid co-linearity problems permitting to work with a number of variables that is greater than the number of samples. A comparison between these two techniques reveals similar results in terms of prediction performance, with no significant difference being reported when both employ the optimised number of principal components (PCs). PLS usually produces good models using a lower number of PCs than its counterpart, PCR. Recent papers comparing four multivariate calibration procedures (including MLR and PLS) employed by NIR technology corroborate the idea that optimised models give similar performances while emphasising that the number of samples employed to construct the model is a relevant problem in the agricultural area.71, 72
For quantitative treatment, it is worth mentioning Artificial Neural Networks (ANN) as an emerging alternative for NIR calibration.73,74 This technique may present some advantages when non-linearity (not easily accommodated by PCR and PLS) between the spectral data and the quantitative information of interest exists.
Considering the synergism between NIR technology and Chemometrics, it is probable that new data treatment alternatives, which can help make good use of the NIR spectroscopic information, will be provided in the near future. Meanwhile, a good review and re-evaluation of existing and even pioneering methods can help to consolidate the technology.58
6. Developing a NIR-Based Analytical Method
6.1. Qualitative applications
Developing an analytical application for identification and classification can be made by adopting one of two possible approaches: supervised or unsupervised methods.56 In the supervised method each spectrum in the spectral set used for training the identification/classification algorithm is attributed to a given class. In the unsupervised method no a priori assumption is made about the number of classes present in the sample set. The algorithm must identify (or help to identify, with the aid of the user) how the number of groups within the samples can be distributed (cluster analysis), classify the samples of the training set and, at same time, provide the model for further classification of unknown samples.
In both methods it is of paramount importance to supply the mathematics with a set of spectra of samples which has all possible variabilities among the samples and within each class that will be found in a future test set. It is also important to identify the instrumental contribution to data dispersion, in order to find where the limits of the prediction capability of the model are.
For supervised methods it is easier to account for in class variability by supplying the classification software in the training set with spectra from a representative number of samples of the same class and providing all possible spectral variability, due to physical, chemical and sample presentations. An external test set can and must be provided for validation of the classification model.
With care paid to the aspects mentioned above, the resulting classification model will probably present a good performance. However, the user must be aware that no classification model is immune to ambiguous results. A good knowledge of the sample set, the limits of the classification algorithm and a conscious evaluation (a graphic observation can help) of the results can contribute to make good use of NIR spectroscopy for qualitative purposes.
6.2. Quantitative applications
Basically the same aspects mentioned above are relevant when aiming at the development of a quantitative application of NIR spectroscopy. NIR spectroscopy is not very sensitive. Most of the quantitative applications are targeted to determine major constituents in the sample. In general, the detection limit is about 0.1% (m/m), although, for some specific applications and under favourable characteristics of the sample matrix and analyte, NIR can reach lower values.75 Some of the practical aspects of NIR calibration have been recently described.76
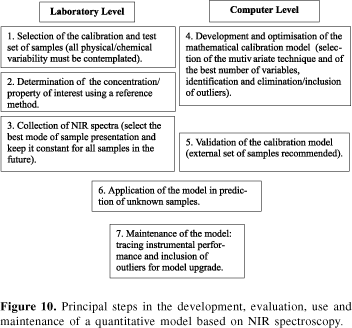
Figure 10 shows the principal steps followed during the development of a quantitative model based on NIR spectroscopy. The user must be aware of the fact that NIR relies on a multivariate model to quantify a property or a concentration in complex samples, such as gasoline, and agricultural products, such as soy beans, wheat flour and sugar cane.
Seldom is it possible to produce a set of artificial calibration samples in the laboratory, an exception is made for very simple samples as, for example, those known to contain only two or three components, such as fuel alcohol, which can be viewed as a binary water-ethanol system.51 Therefore, most of the quantitative models developed by using NIR spectral information are based on the use of samples whose analyte concentration or property has been determined by a standard, well-accepted analytical procedure designated as the reference method. The quality of the results supplied by the reference method (reference results) must be well known, to guide in the validation step of the model.
The samples necessary to produce a robust model must include the natural variability of the concentration or property of interest while a number of samples more-or-less uniformly distributed between the extremes values can give better results. This is not easy to obtain, for example, in products resulting from well-designed industrial processes, because the spread of the property/concentration values will be very small. The minimum range that a property or concentration value must vary in order to provide enough information for the calibration software has been recommended as ± 5 times (and not less than ± 3 times) the reproducibility of the reference method.77
The number of samples employed for calibration has been considered of great importance (perhaps, even more important than the chemometric technique employed for model development). Recommendations for multicomponent natural samples are in the range of 50 to 100 samples, depending on the complexity and variability of the matrix accompanying actual samples. If, for instance, a model is being designed to assess the protein content of wheat grains in general, all types of wheat must be represented in the calibration set; for each class of wheat the concentration of protein must span the expected values. Furthermore, factors affecting the NIR spectrum and those related with seasonal variations must be represented in the sample set. Inclusion of these factors of variability in the model will lead to having the number of samples used for calibration reach 100 or, perhaps, even 1000, depending on sample complexity.
Among the samples supplied for calibration, many may be classified as outliers. These samples do not belong to the majority of the sample population and may reveal a mistake in the concentration/property value obtained using the reference method or may be a sample which was presented to the spectrophotometer in a different way. In addition, and more important, the outlier may be an actual sample presenting some spectral characteristic reflecting a different composition in relation to the majority of the calibration samples. Outliers of such a type should not simply be eliminated. On the contrary, more samples of the same class should be included to account for such variability in composition in order to make the model more robust and of broader application. Criteria and algorithms for outlier detection are supplied with most commercial software.
Any regression algorithm will require a certain number of variables to be included in the final model. These can be original variables (wavelength), as in MLR, or principal components (latent variables), as in PCR and PLS. The best number of variables must be chosen. The procedure for choosing the best number of variables may be preceded by a variable selection algorithm such as described above. In this case, usually, the model has been simultaneously optimised by using an external test set. For PCR and PLS the number of PCs to be included in the model is fundamental. Commercial software packages include automatic optimisation algorithms for suggesting the optimal number of variables. However, it is recommended to the user to closely watch the process and make his own observations and conclusions. Graphical charts showing the behaviour of the amount of explained variability as a function of the number of PCs included in the model can help in this optimisation step. However, one should be aware that an increase in the number of PCs, due their nature, will always decrease the variability not accounted for by the model. Unwanted variability in the data set, such as random noise, may also be taken into account by a model constructed with too many PCs. The model is said to be overfitted, showing excellent results for evaluating samples belonging to the calibration set but failing on prediction of an external validation set. Looking at the differences between the final results (value of the property or concentration) predicted by the model and the reference values and the effect of the number of PCs on this difference is a secure way to optimise the model for the proper number of variables. This can be done by using cross-validation procedures or by using an external set of samples. Cross-validation will remove one sample or a segment of correlated samples from the calibration data set and construct the model without these data. The sample or samples left out then have their values predicted by the model and a calibration error is calculated. The procedure is repeated for each sample or subset of samples and a root mean square error of validation (RMSEV) is computed.
For an external set of samples the performance of the model is usually measured by the root mean square error of prediction (RMSEP), calculated as:

where
i and yi are, respectively, the predicted and reference values of the concentration/property for the external set of Np number of samples.
Validation is the final step in the development of a quantitative model. The use of another external validation set of samples and the RMSEP is recommended because it produces more realistic results which are independent of any data employed in model construction and during the optimisation of its size.
The final model, employed for routine analyses, needs to be periodically checked for performance, along with long term instrumental fluctuations. The detection of outliers must be employed at this stage to check for model extrapolation (a condition in which a multivariate model should not be applied). Intensive use may dictate the necessity of inclusion of more reference samples and running the construction and optimisation steps again to improve the robustness of the model.
Finally, efforts in making a model work with spectral data generated by an instrument other than the one used for its development has become a topic of great importance in NIR spectroscopy. Instrument standardisation or transference of calibration have been the names given to this research topic. Transference of calibration has been the motif of many articles in the literature and the subject appears to be far away from a definitive and unique solution.78,79
7. Applications
It is not surprising, in view of the almost universal information carried by an NIR spectrum, that the NIR technology has found a vast field of application. Table 3 lists some of the sectors where NIR technology has succeed for qualitative and quantitative purposes. The principal attractiveness of the technique, its direct and non-invasive nature, has been the driving force towards its application in agriculture and in process analysis. It is rare to see a NIR-based analytical method requiring more than a physical pre-treatment of the sample (such as grinding and transfer) before spectral data acquisition. Therefore, an analytical method based on NIR spectroscopy offers the rapid response required for in-line and intensive and even for "100% quality control" applications.
The agricultural area was the first to make intensive use of NIR spectroscopy. In fact, this area was responsible for launching the technology on the market and for starting its development up to the standard it has reached today. The most recent use of NIR technology in this sector has been in supporting precision agriculture. The possibility of using NIR instruments in the crop field, collecting data in real time and correlating it with soil composition, is essential to produce the refined data necessary for precision agriculture. Following agricultural applications, many others areas are now experiencing an intensive use of NIR technology. The pharmaceutical and petrochemical industries (including the polymer segment) are the areas where NIR infrared spectroscopy has recently encountered large applicability.
NIR image, clinical optical tomography and in-vivo, non-invasive biochemical analysis, are fields where many research efforts have been focused in recent years, showing promising results.
Table 3 also presents some selected references to a number of articles representative of applications. It is not an exhaustive survey about the contributions to each field. Instead, these references aim at illustrating the potentiality of the NIR technology. Many of the cited articles are reviews on application of NIR spectroscopy in the respective sectors. The Brazilian contributions to each field have been included among many international references, to illustrate the fields where Brazilian researchers have produced some results regarding applied NIR spectroscopy.
8. NIR Spectroscopy in Brazil
A survey made with the aid of the ISI (Institute for Scientific Information) "Webofscience" data base revealed a total of only about 30 contributions to the field of NIR technology published up to September 2002 involving Brazilian researchers working in Brazilian institutions. The Brazilian scientific production is, therefore, very small, mainly when compared with the reported 15000 international contributions to the field.
In relation to the sectors of application of NIR spectroscopy listed in Table 3, the Brazilian contributions concentrate in two of them. The agricultural area was the first, in 1991, to have scientific papers20,21 dealing with the development of analytical methodologies for the determination of natural products. Perhaps this was a consequence of the great international impact this sector experimented as a result of the unique non-destructive and non-invasive approach to analysis of agricultural and food products when using NIR technology. Other publications, in the last four years, reveal other applications in the agricultural area86-90,98,102-105 with papers on zootechnology,86-89 eucalyptus pulp analysis,90 and organic matter determination102,103 and nitrogen content determination105 in soils. Many of these papers show advancements in data analysis and information extraction, improving the Chemometrics employed for NIR calibration.102,104,105 In fact, Brazil has shown the expertise necessary for suitable treatment of data generated by NIR spectroscopy with many groups in activity all over Brazil, employing and/or developing Chemometric methodologies.
The polymer sector appears as the second most contemplated by Brazilian researchers. Articles on polymer processing112-115 and on polymer characterisation122, 123 are all recent contributions and reveal the interest in the application of NIR spectroscopy in this sector, mainly for process control.
The petroleum and fuel industry has received some attention, although only few papers have been published. Research has been developed aimed at the specific characteristics of the Brazilian fuel matrix, which makes use of ethanol as an additive or uses hydrated ethanol directly as fuel.51,63 Although only one paper has been published so far on the determination of quality parameters related to fuel samples,125 it is known, as a function of a broad national effort promoted by the Brazilian Petroleum Agency (ANP) and FINEP (Brazilian agency for financial support of research and development), that many research groups around the country are now involved in the development of NIR-based analytical methods to replace cumbersome and expensive conventional methods for the routine quality control of combustibles.
Some contributions to the field of biomedical and clinical analysis are also found in the literature.149-151 Most of them are, in fact, related to the use of Raman near-infrared spectroscopy for clinical analysis.149, 150 The pharmaceutical sector shows only one paper concerned with the use of NIR spectroscopy for identification of counterfeit drugs, a problem of social interest in Brazil.164
The academic contribution to the development of NIR instrumentation is concentrated in only two papers describing a Hadamard multiplexed dual-beam spectrophotometer41 and a dual beam AOTF-based instrument that makes use of two monochromatic beams generated by an acousto-optic device for reference and measurement.35 The first commercial Brazilian NIR spectrophotometer was launched in 1999 by FEMTO Ltda. and it is based on conventional, low cost dispersive optics. Nevertheless, the company is investing in the AOTF technology, with support of FINEP, and soon a NIR-AOTF-based spectrophotometer will become available. The interest of Brazilian companies and representatives in commercialising and producing NIR instrumentation can only have a positive effect on diffusion of the technology in the near future.
Brazilian expertise in NIR spectroscopy is, not unexpectedly, concentrated in the south-east of the country with the Chemistry Institute of the State University of Campinas in state of São Paulo being responsible for the most significant part of the Brazilian scientific production in NIR spectroscopy applied to Analytical Chemistry. However, this picture is going to undergo significant changes in the near future when many projects in execution throughout the country begin to produce results. The realisation of the first workshop in NIR spectroscopy in 2001 contributed to unite the Brazilian research community and to promote its contact with international experts and industrial representatives. The multiplicative effect of this event will start to be noticed with the number of courses, publications and research projects augmenting significantly.
An overview of the Brazilian contribution to the NIR spectroscopy reveals that the majority of the research papers have been published within the last four years. This concentration allows to predict a substantial increase of Brazilian scientific production in the field in the near future. This will be welcomed because the country, with an economy presently based on agricultural while developing different industrial activities, presents a high potential for absorbing NIR technology in many sectors awaiting development and competitiveness on the international scene. Meanwhile, the role of the universities may be focused on the creation of graduate and undergraduate courses and on the formation of people with the skills required for using NIR technology. This should include a solid foundation in chemometrics, vibrational spectroscopy and modern instrumental Analytical Chemistry. In addition, the exchange of knowledge with the productive sector would help the diffusion of NIR technology, with a synergetic effect on the development of NIR-based analytical methods of national interest.
Conclusions
Undoubtedly, NIR spectroscopy is playing and will play a central role in permitting many industrial and productive sectors to conform to modern high quality standard requirements. However, it is not a panacea and presents some limitations. Some of them are related to its nature as a secondary method. This means that a conventional, well accepted supporting (reference) method must be available to supply the analytical results required for the modelling step of NIR spectral data. The model may be very complicated and is usually resistant to chemical interpretation. Variable selection algorithms have succeeded in diminishing these negative aspects. Furthermore, the models need to be frequently updated to accommodate changes in the sample matrix, even for the same type of sample and analyte. Robust models may require hundreds or even thousands of samples pre-analysed by the reference method. The desired prompt transference of the model to other instruments, capable of attenuating the cost of model construction, is still a matter under study and development. Finally, the technique is not very sensitive; the limits of determination are in the tenths of per cent range.
On the other hand, the universal nature of the information that NIR spectroscopy generates, the non-invasive and non-destructive use allowed by the technique, its expeditiousness, and the robustness of the NIR spectrophotometers commercially available today may overcome the disadvantages indicated herein. The number of scientific papers and the successes of international congresses on the theme are evidence of this fact.
NIR technology has been, for more than four decades, able to incorporate all the benefits brought by the evolution of correlated fields such as Chemometrics, new materials for optical components, new sensors and sensor arrays, microcomputers and micro-electronics. Undoubtedly, NIR has come to stay and its ability of quickly incorporating the advances in the correlated fields ensures it an extensive and fruitful field for research and development.
Acknowledgements
The author is grateful to Dr. Carol H. Collins for manuscript revision and FINEP/CTPETRO/ANP for financial supporting.
References
1. Williams, P.; Norris, K., eds.; Near-Infrared Technology, 2nd ed. , American Association of Cereal Chemistry, Inc.: St. Paul, MN, USA, 2001.
2. Herschel, W.; Phil Trans. Roy. Soc. London 1800, Part II, 255.
3. Herschel, W.; Phil Trans. Roy. Soc. London, 1800, Part II, 284.
4. Wetzel, D.L.; Anal. Chem. 1983, 55, 1165A.
5. Burns, D.A; Ciurczak, E.W., eds.; Handbook of Near-Infrared Analysis, Marcel Dekker, Inc.: New York, 1992.
6. Mcclure W.F.; Anal. Chem. 1994, 66, A43.
7. Davies, T.; Analusis 1998, 26, M17
8. Ellis, J.; Bath, J.; J. Chem. Phys. 1938, 6, 723.
9. Barchewitz, P.; J. Chem. Phys. 1943, 45, 40.
10. Evans, A.; Hibbard, R.R.; Powell A.S.; Anal. Chem. 1951, 23, 1604.
11. White Jr., L.; Barrett, W.J.; Anal. Chem. 1956, 28, 1538.
12. Whetsel, K.; Roberson, W.E.; Krell, M.W.; Anal. Chem. 1958, 30, 1594.
13. Kubelka, P.; Munk, F.; Zeit. Tech. Physik. 1931, 12, 593.
14. Hart, J.R.; Norris, K.H.; Golumbic, C.; Cereal Chem. 1961, 39, 94.
15. Proceedings of the 1963 International Symposium on Humidity and Moisture, Principles and Methods of Measuring Moisture in Liquid and Solids, vol. 4, Reinhold Publishing Co., New York, 1965, p.19.
16. Hart, J.H.; Norris, K.H.; J. Near Infrared Spectrosc. 1996, 4, 23.
17. Ben-Gera, I.; Norris, K.; J. Feed Sci. 1968, 33, 64.
18. Kowalski, B.R.; Anal. Chem. 1980, 52, R112.
19. Blanco, M.; Villarroya, I.; Trends Anal. Chem. 2002, 21, 240.
20. Nishiyama, P.; Alvarez, M.; Vieira, L.G.E.; Arq. Biol. Tecnol. 1991, 34, 361.
21. Nishiyama, P.; Alvarez, M.; Vieira, L.G.E.; J. Sci. Food Agr. 1992, 59, 277.
22. Weyer, L.G.; Appl. Spectrosc. Rev. 1985, 21, 1.
23. Workman Jr., J.J.; Appl. Spectrosc. Rev. 1996, 31, 251.
24. Bokobza, L.; J. Near Infrared Spectrosc. 1998, 6, 3.
25. Scott, D.M.; Meas. Sci. 1995, 6, 156.
26. Morimoto, S.; McClure, W.F.; Stanfield, D.L.; Appl. Spectrosc. 2001, 55, 182.
27. Saito, M.; Furukaua, H.; Appl. Phys. Lett. 2001, 79, 4283.
28. Green, A.E.; Kohsiek, W.; Boundary-layer Metereology 1995, 74, 353.
29. Schnable, J.G.; Grochowski, P.J.; Wilhelm, L.; Harding, C.; Kiefer, M.; Orr, R.S.; Field Anal. Chem. Technol. 1998, 2, 21.
30. McClure, W.F.; Moody, D.; Stanfield, D.L.; Kinoshita, O.; Appl. Spectrosc. 2002, 56, 720.
31. Wilson, A.C.; Barnes, T.H.; Seakins, P.J.; Rolfe, T.G.; Meyer, E.J.; Rev. Sci. Instrum. 1995, 66, 5618.
32. Malinen, J.; Kansakoski, M.; Rikola, R.; Eddison, C.G.; Sens. Actuac. B. 1998, 51, 220.
33. Tran, C.D.; Anal. Chem. 1992, 64, 971A.
34. Xu, J.; Stroud, R.; Acousto-Optic Devices, John Wiley & Sons Inc.: New York, 1992.
35. Guchardi, R.; Pasquini, C.; Appl. Spectrosc. 2001, 55, 454.
36. Barton II, F.E.; Spectrosc. Eur. 2002, 14, 12.
37. Haeusler, A.; Rode, M.; Paul, C. In Near Infrared Spectroscopy: Proceedings of the 10th International Conference, Davies, A.M.C.; Cho, R.K. eds., NIR Publications: Chischester, 2002, p.345.
38. Tilotta, D.C.; Hammaker, R.M.; Fateley, W.G.; Appl. Spectrosc. 1987, 41, 727.
39. Treado, P.J.; Morris, M.D.; Anal. Chem. 1989, 61, 723A.
40. Winefordner, J.D.; Fitzgerald, J.J.; Omenetto, N.; Appl. Spectrosc., 1975, 29, 369.
41. da Silva H.E.B.; Pasquini C.; Appl. Spectrosc. 2001, 55, 715.
42. Turner, II, J.F.; Treado, P.J.; Appl. Spectrosc. 1996, 50, 277.
43. Mortensen, A.N.; Dyer, S.A.; Hammaker, R.M.; Fateley, W.G.; IEEE Trans. Instrumen. Meas. 1996, 45, 394.
44. Hendra, P.J.; Internet J. Vibr. Spectrosc. 2001, 5, [www.ijvs.com].
45. Johanson, J.; Folestad, S.; Josefson, M.; Sparen, A.; Abrahamsson, C.; Ansersson-Engels, S.; Svanberg, S.; Appl. Spectrosc. 2002, 56, 725.
46. Kawano, S. In Near Infrared Spectroscopy: Proceedings of the 10th International Conference, Davies, A.M.C. ; Cho, R.K. eds., NIR Publications: Chischester, 2002, p.15.
47. Miller, C.E. In Near-Infrared Technology ; Williams, P.; Norris, K. eds., 2nd ed. , American Association of Cereal Chemistry, Inc.: St. Paul, Minnesota, USA, 2001, p. 19.
48. Harrington, P.B.; Aaron, U.; Tandler, P.J.; Chem. Intell. Lab. Syst. 2000, 50, 149.
49. Noda, I.; Dowrey, A.E.; Marcott, C.; Story, G.M.; Ozaki, Y.; Appl. Spectrosc. 2000, 54, 236A.
50. Lin, J.; Brown, C.W.; Appl. Spectrosc. 1993, 47, 239.
51. Pasquini, C.; da Silva, H.E.B.; Guchardi, R. In Near Infrared Spectroscopy: Proceedings of the 9th International Conference; Davies, A.M.C.; Giangiacomo R. eds., NIR Publications: Chichester, 2000, p. 109; Tipparat, P.; Lapanantnoppakun, S.; Jakmunee, J.; Grudpan, K.; Talanta 2001, 53, 1199; Lopezanreus, E.; Garrides, S.; Delaguardia, M.; Fresenius J. Anal. Chem. 1995, 351, 724.
52. Dardene, P.; Sinnaeve, G.; Baeten, V.; J. Near Infrared Spectrosc. 2000, 8, 229.
53. Otto, M.; Chemometrics. Statistics and Computer Application in Analytical Chemistry, Wiley-VCH: New York, 1998.
54. Naes, T.; Isaksson, T.; Fearn, T.; Davies, T.; Multivariate Calibration and Classification, NIR Publications: Chichester, 2002.
55. Mark H.; Anal. Chim. Acta 1989, 223, 75.
56. Downey, G.; Analyst 1994, 119, 2367.
57. Hopkins, D.W.; NIR News 2001, 12, 3.
58. Norris, K.H.; NIR News 2001, 12, 6.
59. Savitzky, A.; Golay, M.J.E.; Anal. Chem. 1964, 36, 1627.
60. Geladi, P.; McDougall, D.; Marten, H.; Appl. Spectrosc. 1985, 39, 491.
61. Barnes, R.J.; Dhanoa, M.S.; Lister, S.J.; Appl. Spectrosc. 1989, 43, 772.
62. Leardi, R.; Boggia, R.; Terrile, M.; J. Chemometr. 1992, 6, 267.
63. Guchardi, R.; Costa, P.A.; Poppi, R.J.; Pasquini, C.; J. Near Infrared Spectrosc. 1998, 6, 333.
64. Westad, F.; Martens, H.; J. Near Infrared Spectrosc. 2000, 8, 117.
65. Lundsberg-Nielsen, L.; Kornbo, C.; Bruhn, M.; Dyrby, M. In Near Infrared Spectroscopy: Proceedings of the 10th International Conference; Davies, A.M.C.; Cho, R.K. eds., NIR Publications: Chichester, 2002, p.485.
66. Dreassi, E.; Ceramelli, G., Corti, P.; Perruccio, P.L.; Lonardi, S.; Analyst 1996, 121, 219.
67. Gerhausser, C.; Kovar, K.A.; Appl. Spectrosc. 1997, 51, 1504.
68. Blanco, M.; Romero, W.A.; Analyst 2001, 126, 2212.
69. European Pharmacopoeia, 3rd ed., Council of Europe: Strasbourg, 1997.
70. United States Pharmacopoeia, USP-24-NF 19, United States Pharmacopeial Convention: Rockville, 2000.
71. Dardenne, P.; Sinnaeve, G.; Baeten, V.; J. Near Infrared Spectrosc. 2000, 8, 229.
72. Huang, J.; Brennan, D.; Sattler, L.; Alderman, J.; Lane, B.; O'Mathuna, C.; Chem. Intell. Lab. System. 2002, 62, 25.
73. Zupan, J.; Gasteiger, J.; Neural Networks in Chemistry and Drug Design, 2nd ed., Wiley-VCH: New York, 1999.
74. Brown, C.W.; Anal. Chem. 1998, 70, 2983.
75. Baptista, M.S.; Tran, C.D.; Gao, G.H.; Anal. Chem. 1996, 68, 971.
76. Widemann, S.C.C.; Hansen, W.G.; Snieder, M.; Wortel, V.A.L.; Analusis 1998, 26, M38.
77. ASTM E1655, Standard Practices for Infrared Multivariate Quantitative Analysis, 2000.
78. Dreassi, E.; Ceramelli, G.; Perruccio, P.L.; Corti, P.; Analyst 1998, 123, 1259.
79. Fearn, T.; J. Near Infrared Spectrosc. 2001, 9, 229.
80. Cozzolino, D.; de Mattos, D.; Vaz Martins, D.; Animal Sci. 2002, 74, 477.
81. Davies, A.M.C.; Radovic B.; Fearn T.; Anklam, E.; J. Near Infrared Spectrosc. 2002, 10, 121.
82. Roggo, Y.; Duponchel, L.; Noe, B.; Huvenne, J.P.; J. Near Infrared Spectrosc. 2002, 10, 137.
83. Manley, M.; Van Zyl, L.; Osborne, B.G.; J. Near Infrared Spectrosc. 2002, 10, 71.
84. Tarkosova, J.; Copikova, J.; J. Near Infrared Spectrosc. 2000, 8, 21.
85. Garcia-Alvarez, M.; Huidobro, J.F.; Hermida, M.; Rodriguez-Otero, J.L.; J. Agr. Food Chem. 2000, 48, 5154.
86. Leite, E.R.; Stuth, J.W.; J. Range Manage. 1994, 47, 312.
87. Fonseca, C.E.L.; Hansen, J.L.; Thomas, E.M.; Pell, A.N.; Viands, D.R.; Crop. Sci. 1999, 39, 1265.
88. Leite, E.R.; Stuth, J.W.; Small Ruminant Res. 1995, 15, 223.
89. Pirea, F.F.; Prates, E.R.; Rev. Bras. Zootecn. 1998, 27, 1076.
90. Fardim, P.; Ferreira, M.M.C.; Duran, N.; J. Wood Chem. Technol. 2002, 22, 67.
91. Twomey, M.; Downey, G; McNulty, P.B.; J. Sci. Food Agric. 1995, 67, 77.
92. Isaksson, T.; Naerbo, G.; Rukke, E.O.; Sahni, N.S.; J. Near Infrared Spectrosc. 2001, 9, 11.
93. Kays, S.E.; Barton, F.E.; Windham, W.R.; J. Near Infrared Spectrosc. 2000, 8, 35.
94. Hoyer, H.; Process Control Qual., 1997, 9, 143.
95. Buning-Pfaue, H.; Hartmann, R.; Harder, J.; Kehraus, S.; Urban, C.; Fresenius. 1998, 360, 832.
96. McQueen, D.H.; Wilson, R.; Kinnunen, A.; Jensen, E.P.; Talanta 1995, 42, 2007.
97. Wilson, T.E.; Int. Sugar J. 1996, 98, 169-& .
98. Barthus, R.C.; Poppi, R.J.; Vibr. Spectrosc. 2001, 26, 99.
99. Confalonieri, M.; Fornasier, F.; Ursino, A.; Boccardi, F.; Pintus, B.; Odoardi M.; J. Near Infrared Spectrosc. 2001, 9, 123.
100. Reeves, J.B.; McCarty, G.W.; Meisinger, J.J.; J. Near Infrared Spectrosc. 1999, 7, 179.
101. Stenberg. B.; Jonsson, A.; Borjesson, T. In Near Infrared Spectroscopy: Proceedings of the 10th International Conference; Davies, A.M.C.; Cho, R.K.eds., NIR Publications: Chichester, 2002, p.279.
102. Fidencio, P.H.; Poppi, R.J.; de Andrade, J.C.; Anal. Chim. Acta 2002, 453, 125.
103. Fidencio, P.H.; Poppi, R.J.; de Andrade, J.C.; Cantarella, H.; Commun. Soil Sci. Plant Anal. 2002, 33, 1607.
104. Fidencio, P.H.; Ruisanchez, I.; Poppi, R.J.; Analyst 2001, 126, 2194.
105. Mello, C.; Poppi, R.J.; de Andrade, J.C.; Cantarella, H.; Analyst 1999, 124, 1669.
106. Pasikatan, M.C.; Steele, J.L.; Spillman, K.; Haque, E.; J. Near Infrared Spectrosc. 2001, 9, 153.
107. Wu, P.; Siesler, H.W.; J. Near Infrared Spectrosc. 1999, 7, 65.
108. Lousberg, H.H.A.; Boelens, H.F.M.; Le Comte, E.P.; Hoefsloot, H.C.J.; Smilde, A.K.; J. Appl. Polym. Sci. 2002, 84, 90.
109. Camacho, W.; Karlsson, S.; Polym. Eng. Sci. 2001, 41,1626.
110. Rohe, T.; Becker, W.; Kolle, S.; Eisenreich, N.; Eyerer, P.; Talanta 1999, 50, 283.
111. Fischer, D.; Eichhorn, K.J.; Analusis 1998, 26, M58.
112. Santos A.F.; Lima, E.L.; Pinto, J.C.; J. Appl. Polym. Sci. 1998, 70, 1737.
113. Santos, A.F.; Lima, E.L.; Pinto, J.C.; J. Appl. Polym. Sci. 2000, 77, 453.
114. Vieira, R.A.M.; Sayer, C.; Lima, E.L.; Pinto, J.C.; J. Appl. Polym. Sci. 2001, 84, 2670.
115. Vieira, R.A.M.; Sayer, C.; Lima, E.L.; Pinto, J.C.; Ind. Eng. Chem. Res. 2002, 41, 2915.
116. Shimoyama, M.; Matsukawa, K.; Inoue, H.; Ninomiya, T.; Ozaki, Y.; J. Near Infrared Spectrosc. 1999, 7, 27.
117. Lachenal, G.; Ozaki, Y.; Macromol. Symp. 1999, 141, 283.
118. Bledzki, A.K.; Kardasz, D.; Polimery-W, 1998 43, 79.
119. Lachenal, G.; Int. J. Polym. Anal. Ch. 1997, 3, 145.
120. Camacho, W.; Karlsson. S.; Int. J. Polym. Anal. Ch. 2002, 7, 41.
121. Miller, C.E.; Appl. Spectrosc. 1993, 47, 222.
122. Vieira, R.A.M.; Sayer, C.; Lima, E.L.; Pinto, J.C.; Polymer 2001, 42, 8901.
123. De Araújo, S.C.; Kawano, Y.; J. Appl. Polym. Sci. 2002, 85, 199.
124. Hoffmann, U.; Zanier-Szydlowski, N.; J. Near Infrared Sectrosc. 1999, 7, 33.
125. Reboucas, M.V.; Neto, B.D.; J. Near Infrared Spectrosc. 2001, 9, 263.
126. Wang, Z.M.; Hua, W.Y.; Wei, Z.K.; Zhang, H.H.; Wu, H.Z.; Spectrosc. Spect. Anal. 1999, 19, 684.
127. Workman Jr., J.J.; J. Near Infrared Spectrosc. 1996, 4, 69.
128. Kelly, J.J.; Barlow, C.H.; Jinguri, T.M.; Callis, J.B.; Anal. Chem. 1989, 61, 313.
129. Buttner, G.; Process Contr. Qual. 1997, 9, 197.
130. Chung, H.; Ku, M.S.; Appl. Spectrosc. 2000, 54, 239.
131. Knothe, G.; J. Am. Oil Chem. Soc. 2001, 78, 1025.
132. Lambert, D.; Descales, B.; Espinosa, A.; Sanchez, M.; Osta, S.; Gil, J.; Martens, A.; Valleur, M.; Analusis 1995, 23, M20.
133. Valleur, M.; Petrol. Technol. 1999, 4, 81.
134. Hidajat, K.; Chong S.M.; J. Near Infrared Spectrosc. 2000, 8, 53,
135. Blanco, M.; Maspoch, S.; Villarroya, I.; Peralta, X.; Gonzalez, J.M.;Torres, J.; Appl. Spectrosc. 2001, 55, 834.
136. Geladi, P.; Barring, H.; Dabakk, E.; Trygg, J.; Antti, H.; Wold, S.; Karlberg, B.; J. Near Infrared Spectrosc. 1999, 7, 251.
137. Malley, D.F.; Hunter, K.N.; Webster, G.R.B.; J. Soil Contam. 1999, 8, 481.
138. Malley, D.F.In Near Infrared Spectroscopy: Proceedings of the 10th International Conference, Davies, A.M.C.; Cho, R.K. eds., NIR Publications: Chichester, 2002, p.391,
139. Cleve, E.; Bach, E.; Schollmeyer, E.; Anal. Chim. Acta 2000, 420, 163.
140. Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S.; Pages, J.; Anal. Chim. Acta 1999, 383, 291.
141. Church, J.S.; O'Neill, J.A.; Woodhead, A.L.; Appl. Spectrosc. 1998, 52, 1039.
142. Wang, J.; Geng, Y.J.; Guo, B.; Klima, T.; Lal, B.N.; Willerson, J.T.; Casscells, W.; J. Am. Coll. Cardiol. 2002, 39, 1305.
143. Hazen, K.H.; Arnold, M.A.; Small, G.W.; Anal. Chim. Acta 1998, 371, 255.
144. Fendel, S.; Schrader, B.; Fresenius J. Anal. Chem. 1998, 360, 609.
145. Keller, E.; Ishihara, H.; Nadler, A.; Niederer, P.; Seifert, B.; Yonekawa, Y.; Frei, K.; J. Neurosci. Meth. 2002, 117, 23.
146. Tamura, M.; Hoshi, Y.; Okada, F.; Philos. Trans. Roy. Soc. B 1997, 352, 737.
147. Malin, S.F.; Ruchti, T.L.; Blank, T.B.; Thennadil, S.N.; Monfre, S.L.; Clin. Chem. 1999, 45, 1651.
148. Daneshvar, M.I.; Peralta, J.M.; Casay, G.A.; Narayanan, N.; Evans, L.; Patonay, G.; Strekowski, L.; J. Immunol. Method. 1999, 226, 119.
149. Duarte, J.; Pacheco, M.T.T.; Machado, R.Z.; Silveira, L.; Zangaro, R.A.; Villaverde, A.B.; Cell Mol. Biol. 2002, 48, 585.
150. Pilotto, S.; Pacheco, M.T.T.; Silveira, L.; Villaverde, A.B.; Zangaro, R.A.; Laser Med. Sci. 2001, 16, 2.
151. da Costa, P.A.; Poppi, R.J.; Anal. Chim. Acta 2001, 446, 39.
152. Sparen, A.; Malm, M.; Josefson, M.; Folestad, S.; Johansson, J.; Appl. Spectrosc. 2002, 56, 586.
153. Kramer, K.; Ebel, S.; Anal. Chim. Acta 2000, 420, 155.
154. Aldrige, P.K.; Mushinsky, R.F.; Andino, M.M.; Evans, C.L.; Appl. Spectrosc. 1994, 48, 1272.
155. Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S.; de la Pezuela, C.; Analyst 1998, 123, 135R.
156. Blanco, M.; Coello, J.; Eustaquio, A.; Iturriaga, H.; Maspoch, S.; Anal. Chim. Acta 1999, 392, 237.
157. Blanco, M.; Romero, M.A.; Analyst 2001, 126, 2212.
158. El-Hagrasy, A.S.; Morris, H.R.; D'Amico, F.; Lodder, R.A.; Drennen, J.K.; J. Pharm. Sci. 2001, 90, 1298.
159. Borer, M.W.; Zhou, X.J.; Hays, D.M.; Hofer, J.D.; White, K.C.; J. Pharmaceut. Biomed. 1998, 17, 641.
160. Martin, K.; Appl. Spectrosc. 1998, 52, 1001.
161. Pande, C.M.; Yang, B.; J. Cosmet. Sci. 2000, 51, 183.
162. Woo, Y.A.; Ahn, J.W.; Chun, I.K.; Kim, H.J.; Anal. Chem. 2001, 73, 4964.
163. Trafford, A.D.; Jee, R.D.; Moffat, A.C.; Graham, P.; Analyst 1999, 124, 163.
164. Scafi, S.H.F.; Pasquini, C.; Analyst 2001, 126, 2218.
165. Tran, C.D.; J. Near Infrared Spectrosc. 2000, 8, 87.
166. Martinsen, P.; Schaare, P.; Andrews, M.; J. Near Infrared Spectrosc. 1999, 7, 17.
167. Tran, C.D.; Politi, M.; Anal. Chem. 2002, 74, 1604.
168. Politi, M.; Tran, C.D.; J. Non-Cryst. Solids 2002, 304, 64.
Received: October 19, 2002
Published on the web: April 7, 2003
FAPESP helped in meeting the publication costs of this article
- 1. Williams, P.; Norris, K., eds.; Near-Infrared Technology, 2nd ed. , American Association of Cereal Chemistry, Inc.: St. Paul, MN, USA, 2001.
- 2. Herschel, W.; Phil Trans. Roy. Soc. London 1800, Part II, 255.
- 3. Herschel, W.; Phil Trans. Roy. Soc. London, 1800, Part II, 284.
- 4. Wetzel, D.L.; Anal. Chem. 1983, 55, 1165A.
- 5. Burns, D.A; Ciurczak, E.W., eds.; Handbook of Near-Infrared Analysis, Marcel Dekker, Inc.: New York, 1992.
- 6. Mcclure W.F.; Anal. Chem. 1994, 66, A43.
- 7. Davies, T.; Analusis 1998, 26, M17
- 8. Ellis, J.; Bath, J.; J. Chem. Phys. 1938, 6, 723.
- 9. Barchewitz, P.; J. Chem. Phys. 1943, 45, 40.
- 10. Evans, A.; Hibbard, R.R.; Powell A.S.; Anal. Chem. 1951, 23, 1604.
- 11. White Jr., L.; Barrett, W.J.; Anal. Chem. 1956, 28, 1538.
- 12. Whetsel, K.; Roberson, W.E.; Krell, M.W.; Anal. Chem. 1958, 30, 1594.
- 13. Kubelka, P.; Munk, F.; Zeit. Tech. Physik. 1931, 12, 593.
- 14. Hart, J.R.; Norris, K.H.; Golumbic, C.; Cereal Chem. 1961, 39, 94.
- 15. Proceedings of the 1963 International Symposium on Humidity and Moisture, Principles and Methods of Measuring Moisture in Liquid and Solids, vol. 4, Reinhold Publishing Co., New York, 1965, p.19.
- 16. Hart, J.H.; Norris, K.H.; J. Near Infrared Spectrosc 1996, 4, 23.
- 17. Ben-Gera, I.; Norris, K.; J. Feed Sci. 1968, 33, 64.
- 18. Kowalski, B.R.; Anal. Chem. 1980, 52, R112.
- 19. Blanco, M.; Villarroya, I.; Trends Anal. Chem. 2002, 21, 240.
- 20. Nishiyama, P.; Alvarez, M.; Vieira, L.G.E.; Arq. Biol. Tecnol. 1991, 34, 361.
- 21. Nishiyama, P.; Alvarez, M.; Vieira, L.G.E.; J. Sci. Food Agr. 1992, 59, 277.
- 22. Weyer, L.G.; Appl. Spectrosc. Rev. 1985, 21, 1.
- 23. Workman Jr., J.J.; Appl. Spectrosc. Rev. 1996, 31, 251.
- 24. Bokobza, L.; J. Near Infrared Spectrosc. 1998, 6, 3.
- 25. Scott, D.M.; Meas. Sci. 1995, 6, 156.
- 26. Morimoto, S.; McClure, W.F.; Stanfield, D.L.; Appl. Spectrosc 2001, 55, 182.
- 27. Saito, M.; Furukaua, H.; Appl. Phys. Lett. 2001, 79, 4283.
- 28. Green, A.E.; Kohsiek, W.; Boundary-layer Metereology 1995, 74, 353.
- 29. Schnable, J.G.; Grochowski, P.J.; Wilhelm, L.; Harding, C.; Kiefer, M.; Orr, R.S.; Field Anal. Chem. Technol. 1998, 2, 21.
- 30. McClure, W.F.; Moody, D.; Stanfield, D.L.; Kinoshita, O.; Appl. Spectrosc. 2002, 56, 720.
- 31. Wilson, A.C.; Barnes, T.H.; Seakins, P.J.; Rolfe, T.G.; Meyer, E.J.; Rev. Sci. Instrum. 1995, 66, 5618.
- 32. Malinen, J.; Kansakoski, M.; Rikola, R.; Eddison, C.G.; Sens. Actuac. B. 1998, 51, 220.
- 33. Tran, C.D.; Anal. Chem. 1992, 64, 971A.
- 34. Xu, J.; Stroud, R.; Acousto-Optic Devices, John Wiley & Sons Inc.: New York, 1992.
- 35. Guchardi, R.; Pasquini, C.; Appl. Spectrosc. 2001, 55, 454.
- 36. Barton II, F.E.; Spectrosc. Eur. 2002, 14, 12.
- 37. Haeusler, A.; Rode, M.; Paul, C. In Near Infrared Spectroscopy: Proceedings of the 10th International Conference, Davies, A.M.C.; Cho, R.K. eds., NIR Publications: Chischester, 2002, p.345.
- 38. Tilotta, D.C.; Hammaker, R.M.; Fateley, W.G.; Appl. Spectrosc. 1987, 41, 727.
- 39. Treado, P.J.; Morris, M.D.; Anal. Chem. 1989, 61, 723A.
- 40. Winefordner, J.D.; Fitzgerald, J.J.; Omenetto, N.; Appl. Spectrosc., 1975, 29, 369.
- 41. da Silva H.E.B.; Pasquini C.; Appl. Spectrosc. 2001, 55, 715.
- 42. Turner, II, J.F.; Treado, P.J.; Appl. Spectrosc. 1996, 50, 277.
- 43. Mortensen, A.N.; Dyer, S.A.; Hammaker, R.M.; Fateley, W.G.; IEEE Trans. Instrumen. Meas. 1996, 45, 394.
- 44. Hendra, P.J.; Internet J. Vibr. Spectrosc 2001, 5, [www.ijvs.com].
- 45. Johanson, J.; Folestad, S.; Josefson, M.; Sparen, A.; Abrahamsson, C.; Ansersson-Engels, S.; Svanberg, S.; Appl. Spectrosc. 2002, 56, 725.
- 46. Kawano, S. In Near Infrared Spectroscopy: Proceedings of the 10th International Conference, Davies, A.M.C. ; Cho, R.K. eds., NIR Publications: Chischester, 2002, p.15.
- 47. Miller, C.E. In Near-Infrared Technology ; Williams, P.; Norris, K. eds., 2nd ed. , American Association of Cereal Chemistry, Inc.: St. Paul, Minnesota, USA, 2001, p. 19.
- 48. Harrington, P.B.; Aaron, U.; Tandler, P.J.; Chem. Intell. Lab. Syst. 2000, 50, 149.
- 49. Noda, I.; Dowrey, A.E.; Marcott, C.; Story, G.M.; Ozaki, Y.; Appl. Spectrosc. 2000, 54, 236A.
- 50. Lin, J.; Brown, C.W.; Appl. Spectrosc 1993, 47, 239.
- 51. Pasquini, C.; da Silva, H.E.B.; Guchardi, R. In Near Infrared Spectroscopy: Proceedings of the 9th International Conference; Davies, A.M.C.; Giangiacomo R. eds., NIR Publications: Chichester, 2000, p. 109;
- Tipparat, P.; Lapanantnoppakun, S.; Jakmunee, J.; Grudpan, K.; Talanta 2001, 53, 1199;
- Lopezanreus, E.; Garrides, S.; Delaguardia, M.; Fresenius J. Anal. Chem. 1995, 351, 724.
- 52. Dardene, P.; Sinnaeve, G.; Baeten, V.; J. Near Infrared Spectrosc. 2000, 8, 229.
- 53. Otto, M.; Chemometrics. Statistics and Computer Application in Analytical Chemistry, Wiley-VCH: New York, 1998.
- 54. Naes, T.; Isaksson, T.; Fearn, T.; Davies, T.; Multivariate Calibration and Classification, NIR Publications: Chichester, 2002.
- 55. Mark H.; Anal. Chim. Acta 1989, 223, 75.
- 56. Downey, G.; Analyst 1994, 119, 2367.
- 57. Hopkins, D.W.; NIR News 2001, 12, 3.
- 58. Norris, K.H.; NIR News 2001, 12, 6.
- 59. Savitzky, A.; Golay, M.J.E.; Anal. Chem. 1964, 36, 1627.
- 60. Geladi, P.; McDougall, D.; Marten, H.; Appl. Spectrosc. 1985, 39, 491.
- 61. Barnes, R.J.; Dhanoa, M.S.; Lister, S.J.; Appl. Spectrosc. 1989, 43, 772.
- 62. Leardi, R.; Boggia, R.; Terrile, M.; J. Chemometr. 1992, 6, 267.
- 63. Guchardi, R.; Costa, P.A.; Poppi, R.J.; Pasquini, C.; J. Near Infrared Spectrosc. 1998, 6, 333.
- 64. Westad, F.; Martens, H.; J. Near Infrared Spectrosc. 2000, 8, 117.
- 65. Lundsberg-Nielsen, L.; Kornbo, C.; Bruhn, M.; Dyrby, M. In Near Infrared Spectroscopy: Proceedings of the 10th International Conference; Davies, A.M.C.; Cho, R.K. eds., NIR Publications: Chichester, 2002, p.485.
- 66. Dreassi, E.; Ceramelli, G., Corti, P.; Perruccio, P.L.; Lonardi, S.; Analyst 1996, 121, 219.
- 67. Gerhausser, C.; Kovar, K.A.; Appl. Spectrosc. 1997, 51, 1504.
- 68. Blanco, M.; Romero, W.A.; Analyst 2001, 126, 2212.
- 69. European Pharmacopoeia, 3rd ed., Council of Europe: Strasbourg, 1997.
- 70. United States Pharmacopoeia, USP-24-NF 19, United States Pharmacopeial Convention: Rockville, 2000.
- 71. Dardenne, P.; Sinnaeve, G.; Baeten, V.; J. Near Infrared Spectrosc. 2000, 8, 229.
- 72. Huang, J.; Brennan, D.; Sattler, L.; Alderman, J.; Lane, B.; O'Mathuna, C.; Chem. Intell. Lab. System. 2002, 62, 25.
- 73. Zupan, J.; Gasteiger, J.; Neural Networks in Chemistry and Drug Design, 2nd ed., Wiley-VCH: New York, 1999.
- 74. Brown, C.W.; Anal. Chem. 1998, 70, 2983.
- 75. Baptista, M.S.; Tran, C.D.; Gao, G.H.; Anal. Chem. 1996, 68, 971.
- 76. Widemann, S.C.C.; Hansen, W.G.; Snieder, M.; Wortel, V.A.L.; Analusis 1998, 26, M38.
- 77. ASTM E1655, Standard Practices for Infrared Multivariate Quantitative Analysis, 2000.
- 78. Dreassi, E.; Ceramelli, G.; Perruccio, P.L.; Corti, P.; Analyst 1998, 123, 1259.
- 79. Fearn, T.; J. Near Infrared Spectrosc. 2001, 9, 229.
- 80. Cozzolino, D.; de Mattos, D.; Vaz Martins, D.; Animal Sci. 2002, 74, 477.
- 81. Davies, A.M.C.; Radovic B.; Fearn T.; Anklam, E.; J. Near Infrared Spectrosc. 2002, 10, 121.
- 82. Roggo, Y.; Duponchel, L.; Noe, B.; Huvenne, J.P.; J. Near Infrared Spectrosc. 2002, 10, 137.
- 83. Manley, M.; Van Zyl, L.; Osborne, B.G.; J. Near Infrared Spectrosc 2002, 10, 71.
- 84. Tarkosova, J.; Copikova, J.; J. Near Infrared Spectrosc. 2000, 8, 21.
- 85. Garcia-Alvarez, M.; Huidobro, J.F.; Hermida, M.; Rodriguez-Otero, J.L.; J. Agr. Food Chem. 2000, 48, 5154.
- 86. Leite, E.R.; Stuth, J.W.; J. Range Manage. 1994, 47, 312.
- 87. Fonseca, C.E.L.; Hansen, J.L.; Thomas, E.M.; Pell, A.N.; Viands, D.R.; Crop. Sci. 1999, 39, 1265.
- 88. Leite, E.R.; Stuth, J.W.; Small Ruminant Res. 1995, 15, 223.
- 89. Pirea, F.F.; Prates, E.R.; Rev. Bras. Zootecn. 1998, 27, 1076.
- 90. Fardim, P.; Ferreira, M.M.C.; Duran, N.; J. Wood Chem. Technol. 2002, 22, 67.
- 91. Twomey, M.; Downey, G; McNulty, P.B.; J. Sci. Food Agric. 1995, 67, 77.
- 92. Isaksson, T.; Naerbo, G.; Rukke, E.O.; Sahni, N.S.; J. Near Infrared Spectrosc. 2001, 9, 11.
- 93. Kays, S.E.; Barton, F.E.; Windham, W.R.; J. Near Infrared Spectrosc. 2000, 8, 35.
- 94. Hoyer, H.; Process Control Qual., 1997, 9, 143.
- 95. Buning-Pfaue, H.; Hartmann, R.; Harder, J.; Kehraus, S.; Urban, C.; Fresenius 1998, 360, 832.
- 96. McQueen, D.H.; Wilson, R.; Kinnunen, A.; Jensen, E.P.; Talanta 1995, 42, 2007.
- 97. Wilson, T.E.; Int. Sugar J. 1996, 98, 169-&
- 98. Barthus, R.C.; Poppi, R.J.; Vibr. Spectrosc 2001, 26, 99.
- 99. Confalonieri, M.; Fornasier, F.; Ursino, A.; Boccardi, F.; Pintus, B.; Odoardi M.; J. Near Infrared Spectrosc. 2001, 9, 123.
- 100. Reeves, J.B.; McCarty, G.W.; Meisinger, J.J.; J. Near Infrared Spectrosc. 1999, 7, 179.
- 101. Stenberg. B.; Jonsson, A.; Borjesson, T. In Near Infrared Spectroscopy: Proceedings of the 10th International Conference; Davies, A.M.C.; Cho, R.K.eds., NIR Publications: Chichester, 2002, p.279.
- 102. Fidencio, P.H.; Poppi, R.J.; de Andrade, J.C.; Anal. Chim. Acta 2002, 453, 125.
- 103. Fidencio, P.H.; Poppi, R.J.; de Andrade, J.C.; Cantarella, H.; Commun. Soil Sci. Plant Anal. 2002, 33, 1607.
- 104. Fidencio, P.H.; Ruisanchez, I.; Poppi, R.J.; Analyst 2001, 126, 2194.
- 105. Mello, C.; Poppi, R.J.; de Andrade, J.C.; Cantarella, H.; Analyst 1999, 124, 1669.
- 106. Pasikatan, M.C.; Steele, J.L.; Spillman, K.; Haque, E.; J. Near Infrared Spectrosc 2001, 9, 153.
- 107. Wu, P.; Siesler, H.W.; J. Near Infrared Spectrosc. 1999, 7, 65.
- 108. Lousberg, H.H.A.; Boelens, H.F.M.; Le Comte, E.P.; Hoefsloot, H.C.J.; Smilde, A.K.; J. Appl. Polym. Sci. 2002, 84, 90.
- 109. Camacho, W.; Karlsson, S.; Polym. Eng. Sci. 2001, 41,1626.
- 110. Rohe, T.; Becker, W.; Kolle, S.; Eisenreich, N.; Eyerer, P.; Talanta 1999, 50, 283.
- 111. Fischer, D.; Eichhorn, K.J.; Analusis 1998, 26, M58.
- 112. Santos A.F.; Lima, E.L.; Pinto, J.C.; J. Appl. Polym. Sci. 1998, 70, 1737.
- 113. Santos, A.F.; Lima, E.L.; Pinto, J.C.; J. Appl. Polym. Sci. 2000, 77, 453.
- 114. Vieira, R.A.M.; Sayer, C.; Lima, E.L.; Pinto, J.C.; J. Appl. Polym. Sci. 2001, 84, 2670.
- 115. Vieira, R.A.M.; Sayer, C.; Lima, E.L.; Pinto, J.C.; Ind. Eng. Chem. Res. 2002, 41, 2915.
- 116. Shimoyama, M.; Matsukawa, K.; Inoue, H.; Ninomiya, T.; Ozaki, Y.; J. Near Infrared Spectrosc. 1999, 7, 27.
- 117. Lachenal, G.; Ozaki, Y.; Macromol. Symp. 1999, 141, 283.
- 118. Bledzki, A.K.; Kardasz, D.; Polimery-W, 1998 43, 79.
- 119. Lachenal, G.; Int. J. Polym. Anal. Ch. 1997, 3, 145.
- 120. Camacho, W.; Karlsson. S.; Int. J. Polym. Anal. Ch. 2002, 7, 41.
- 121. Miller, C.E.; Appl. Spectrosc 1993, 47, 222.
- 122. Vieira, R.A.M.; Sayer, C.; Lima, E.L.; Pinto, J.C.; Polymer 2001, 42, 8901.
- 123. De Araújo, S.C.; Kawano, Y.; J. Appl. Polym. Sci. 2002, 85, 199.
- 124. Hoffmann, U.; Zanier-Szydlowski, N.; J. Near Infrared Sectrosc. 1999, 7, 33.
- 125. Reboucas, M.V.; Neto, B.D.; J. Near Infrared Spectrosc 2001, 9, 263.
- 126. Wang, Z.M.; Hua, W.Y.; Wei, Z.K.; Zhang, H.H.; Wu, H.Z.; Spectrosc. Spect. Anal. 1999, 19, 684.
- 127. Workman Jr., J.J.; J. Near Infrared Spectrosc. 1996, 4, 69.
- 128. Kelly, J.J.; Barlow, C.H.; Jinguri, T.M.; Callis, J.B.; Anal. Chem. 1989, 61, 313.
- 129. Buttner, G.; Process Contr. Qual. 1997, 9, 197.
- 130. Chung, H.; Ku, M.S.; Appl. Spectrosc. 2000, 54, 239.
- 131. Knothe, G.; J. Am. Oil Chem. Soc. 2001, 78, 1025.
- 132. Lambert, D.; Descales, B.; Espinosa, A.; Sanchez, M.; Osta, S.; Gil, J.; Martens, A.; Valleur, M.; Analusis 1995, 23, M20.
- 133. Valleur, M.; Petrol. Technol. 1999, 4, 81.
- 134. Hidajat, K.; Chong S.M.; J. Near Infrared Spectrosc. 2000, 8, 53,
- 135. Blanco, M.; Maspoch, S.; Villarroya, I.; Peralta, X.; Gonzalez, J.M.;Torres, J.; Appl. Spectrosc. 2001, 55, 834.
- 136. Geladi, P.; Barring, H.; Dabakk, E.; Trygg, J.; Antti, H.; Wold, S.; Karlberg, B.; J. Near Infrared Spectrosc. 1999, 7, 251.
- 137. Malley, D.F.; Hunter, K.N.; Webster, G.R.B.; J. Soil Contam. 1999, 8, 481.
- 138. Malley, D.F.In Near Infrared Spectroscopy: Proceedings of the 10th International Conference, Davies, A.M.C.; Cho, R.K. eds., NIR Publications: Chichester, 2002, p.391,
- 139. Cleve, E.; Bach, E.; Schollmeyer, E.; Anal. Chim. Acta 2000, 420, 163.
- 140. Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S.; Pages, J.; Anal. Chim. Acta 1999, 383, 291.
- 141. Church, J.S.; O'Neill, J.A.; Woodhead, A.L.; Appl. Spectrosc. 1998, 52, 1039.
- 142. Wang, J.; Geng, Y.J.; Guo, B.; Klima, T.; Lal, B.N.; Willerson, J.T.; Casscells, W.; J. Am. Coll. Cardiol. 2002, 39, 1305.
- 143. Hazen, K.H.; Arnold, M.A.; Small, G.W.; Anal. Chim. Acta 1998, 371, 255.
- 144. Fendel, S.; Schrader, B.; Fresenius J. Anal. Chem. 1998, 360, 609.
- 145. Keller, E.; Ishihara, H.; Nadler, A.; Niederer, P.; Seifert, B.; Yonekawa, Y.; Frei, K.; J. Neurosci. Meth. 2002, 117, 23.
- 146. Tamura, M.; Hoshi, Y.; Okada, F.; Philos. Trans. Roy. Soc. B 1997, 352, 737.
- 147. Malin, S.F.; Ruchti, T.L.; Blank, T.B.; Thennadil, S.N.; Monfre, S.L.; Clin. Chem. 1999, 45, 1651.
- 148. Daneshvar, M.I.; Peralta, J.M.; Casay, G.A.; Narayanan, N.; Evans, L.; Patonay, G.; Strekowski, L.; J. Immunol. Method. 1999, 226, 119.
- 149. Duarte, J.; Pacheco, M.T.T.; Machado, R.Z.; Silveira, L.; Zangaro, R.A.; Villaverde, A.B.; Cell Mol. Biol. 2002, 48, 585.
- 150. Pilotto, S.; Pacheco, M.T.T.; Silveira, L.; Villaverde, A.B.; Zangaro, R.A.; Laser Med. Sci. 2001, 16, 2.
- 151. da Costa, P.A.; Poppi, R.J.; Anal. Chim. Acta 2001, 446, 39.
- 152. Sparen, A.; Malm, M.; Josefson, M.; Folestad, S.; Johansson, J.; Appl. Spectrosc. 2002, 56, 586.
- 153. Kramer, K.; Ebel, S.; Anal. Chim. Acta 2000, 420, 155.
- 154. Aldrige, P.K.; Mushinsky, R.F.; Andino, M.M.; Evans, C.L.; Appl. Spectrosc. 1994, 48, 1272.
- 155. Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S.; de la Pezuela, C.; Analyst 1998, 123, 135R.
- 156. Blanco, M.; Coello, J.; Eustaquio, A.; Iturriaga, H.; Maspoch, S.; Anal. Chim. Acta 1999, 392, 237.
- 157. Blanco, M.; Romero, M.A.; Analyst 2001, 126, 2212.
- 158. El-Hagrasy, A.S.; Morris, H.R.; D'Amico, F.; Lodder, R.A.; Drennen, J.K.; J. Pharm. Sci. 2001, 90, 1298.
- 159. Borer, M.W.; Zhou, X.J.; Hays, D.M.; Hofer, J.D.; White, K.C.; J. Pharmaceut. Biomed. 1998, 17, 641.
- 160. Martin, K.; Appl. Spectrosc. 1998, 52, 1001.
- 161. Pande, C.M.; Yang, B.; J. Cosmet. Sci. 2000, 51, 183.
- 162. Woo, Y.A.; Ahn, J.W.; Chun, I.K.; Kim, H.J.; Anal. Chem. 2001, 73, 4964.
- 163. Trafford, A.D.; Jee, R.D.; Moffat, A.C.; Graham, P.; Analyst 1999, 124, 163.
- 164. Scafi, S.H.F.; Pasquini, C.; Analyst 2001, 126, 2218.
- 165. Tran, C.D.; J. Near Infrared Spectrosc. 2000, 8, 87.
- 166. Martinsen, P.; Schaare, P.; Andrews, M.; J. Near Infrared Spectrosc. 1999, 7, 17.
- 167. Tran, C.D.; Politi, M.; Anal. Chem. 2002, 74, 1604.
- 168. Politi, M.; Tran, C.D.; J. Non-Cryst. Solids 2002, 304, 64.
Publication Dates
-
Publication in this collection
13 June 2003 -
Date of issue
Apr 2003
History
-
Accepted
07 Apr 2003 -
Received
19 Oct 2002