Abstracts
Monte Carlo simulations have been performed to investigate the structure and hydrogen bonds formation in liquid acetaldehyde. An all atom model for the acetaldehyde have been optimized in the present work. Theoretical values obtained for heat of vaporisation and density of the liquid are in good agreement with experimental data. Graphics of radial distribution function indicate a well structured liquid compared to other similar dipolar organic liquids. Molecular mechanics minimization in gas phase leads to a trimer of very stable structure. The geometry of this complex is in very good agreement with the rdf. The shortest site-site correlation is between oxygen and the carbonyl hydrogen, suggesting that this correlation play a important role in the liquid structure and properties. The O<FONT FACE=Symbol>×××</FONT>H average distance and the C-H<FONT FACE=Symbol>×××</FONT>O angle obtained are characteristic of weak hydrogen bonds.
Monte Carlo simulation; acetaldehyde; liquid structure; weak hydrogen bond
Investigaram-se a estrutura e formação de pontes de hidrogênio no acetaldeído líquido por simulações de Monte Carlo. Um modelo para acetaldeído com todos os átomos explicitados foi otimizado no presente trabalho. Os valores teóricos obtidos para a entalpia de vaporização e a densidade do líquido mostram boa concordância com os dados experimentais. Os gráficos de função de distribuição radial (rdf) indicam um líquido bem estruturado comparado com outros líquidos orgânicos dipolares semelhantes. A minimização usando mecânica molecular em fase gasosa produz um trímero com uma estrutura muito estável. A geometria deste complexo apresenta boa concordância com os gráficos de rdf. A correlação sítio-sítio mais próxima observada é entre o oxigênio e o hidrogênio da carbonila, sugerindo que esta correlação possui um importante papel na estrutura do líquido e suas propriedades. A distância média O<FONT FACE=Symbol>×××</FONT>H e o ângulo C-H<FONT FACE=Symbol>×××</FONT>O obtidos são característicos de pontes de hidrogênio fracas.
ARTICLE
Structure and weak hydrogen bonds in liquid acetaldehyde
Maria A. M. Cordeiro; João M. M. Cordeiro* * e-mail: cordeiro@fqm.feis.unesp.br
Departamento de Física e Química, Faculdade de Engenharia de Ilha Solteira, Universidade Estadual Paulista, Av. Brasil 56, CP 31, 15.385-000 Ilha Solteira - SP, Brazil
ABSTRACT
Monte Carlo simulations have been performed to investigate the structure and hydrogen bonds formation in liquid acetaldehyde. An all atom model for the acetaldehyde have been optimized in the present work. Theoretical values obtained for heat of vaporisation and density of the liquid are in good agreement with experimental data. Graphics of radial distribution function indicate a well structured liquid compared to other similar dipolar organic liquids. Molecular mechanics minimization in gas phase leads to a trimer of very stable structure. The geometry of this complex is in very good agreement with the rdf. The shortest site-site correlation is between oxygen and the carbonyl hydrogen, suggesting that this correlation play a important role in the liquid structure and properties. The O×××H average distance and the CH×××O angle obtained are characteristic of weak hydrogen bonds.
Keywords: Monte Carlo simulation, acetaldehyde, liquid structure, weak hydrogen bond
RESUMO
Investigaram-se a estrutura e formação de pontes de hidrogênio no acetaldeído líquido por simulações de Monte Carlo. Um modelo para acetaldeído com todos os átomos explicitados foi otimizado no presente trabalho. Os valores teóricos obtidos para a entalpia de vaporização e a densidade do líquido mostram boa concordância com os dados experimentais. Os gráficos de função de distribuição radial (rdf) indicam um líquido bem estruturado comparado com outros líquidos orgânicos dipolares semelhantes. A minimização usando mecânica molecular em fase gasosa produz um trímero com uma estrutura muito estável. A geometria deste complexo apresenta boa concordância com os gráficos de rdf. A correlação sítio-sítio mais próxima observada é entre o oxigênio e o hidrogênio da carbonila, sugerindo que esta correlação possui um importante papel na estrutura do líquido e suas propriedades. A distância média O×××H e o ângulo CH×××O obtidos são característicos de pontes de hidrogênio fracas.
Introduction
In recent years, theoretical investigation of liquid state properties has become a challenging subject for theoretical chemists and physicists, since fast and high memory capability computers have become available at relatively low cost.1-3 The development of suitable computer simulation methods have made it feasible to investigate a great number of liquid state properties that would not be possible, or difficult to access, with other theoretic or experimental techniques.4,5 However, the success of the investigations using computational methodologies depends strongly on the availability of potential functions capable of describing the intermolecular interactions adequately.6-9 Therefore, the optimisation of meaningful intermolecular potential functions is of fundamental importance for the investigation of liquid state properties using theoretical methods. This optimisation is not simple, however, due to the complexity of the liquid intermolecular interactions, and some criteria, such as pairwise additivity and consideration of dipolar interaction exclusively, have been used to simplify the interaction potential making the work feasible.10,11
We have recently contributed to the development of parameterised potential functions to study dipolar organic liquids, using these approximations.12,13 Results on thermodynamics and structural aspects have been published.12-15 The study has been centred in amides, since these molecules are suitable models for peptide moieties, and dimethyl sulfoxide because these liquids are popular solvents in organic chemistry with stressed effects in reaction kinetics.16 In this work, new additive pairwise potential has been parameterised for acetaldehyde. These results are part of a more extensive work aiming to investigate hydrogen bonding in biologically relevant systems, and extend the data earlier published.12,14 In a future paper, thermodynamics results for aqueous solutions of acetaldehyde, formamide and methylformamide will be reported. The results presented here were obtained by Monte Carlo simulation,17 which, along with molecular dynamics,18 is a very popular computational method in this area.19-21 Gas phase molecular mechanics (MM) have been used to improve the insights about the species prevailing in the liquid phase.
Methodology
The intermolecular potential function
Following usual procedures in force field calculations,22 the molecules were modelled by collections of interacting sites. The energy Eab between molecules a and b was given by a sum of electrostatic Coulomb plus non-bonded Lennard-Jones terms centred on the sites:

where rij is the distance between site i in a and site j in b and qi and qj are fractional point charges located on the i and j molecular sites. For each site k, the parameters Akk and Bkk were given by Akk=4eksk12 and Bkk=4eksk6, where ek and sk are the Lennard-Jones parameters for the k-th site. In most of the systems (mainly in the case of polarizable species), many-body terms can play an important role in the interaction potential. However, despite the weight of these terms in the potential, they are rarely included in the computer simulations, for this would be very expensive. The average many-body effects can be partially included in the pairwise approximation by defining an effective pair potential.
Parameters Aij and Bij for a nondiagonal interaction [ i,j] were obtained using the geometric combining rules23Aij = (Aii Ajj)1/2 and Bij = (Bii Bjj)1/2. This is a standard procedure used in this field of study. In an earlier work it was shown that results obtained using the combining rule above and the equally well-known Lorentz-Berthelot rule are equivalent.24
Molecular model and potential parameters
The acetaldehyde optimised in this work is a rigid all-atom model, the geometry of which was taken from all-atom OPLS model.25 Point charges needed to take into account the Coulomb interaction in equation (1) were obtained from Mulliken charges calculated at MP2 level with a 6-31G* basis set. The calculations were performed with the Gaussian 94 program.26 The utilisation of CHELPG procedure27 at the RHF level to calculate the point charges has been suggested in an earlier work.28 Extensive studies have revealed, however, that for molecules with a carbonyl group, mainly those presenting peptide bond, Mulliken charges better represent the molecular charge asymmetry than the CHELPG procedure.29 Mulliken population analysis was used to parameterise the atom partial charges in CHARMM program.9 It has also been suggested that charges obtained using CHELPG are not dependent on the inclusion of electron correlation.30 However, previous calculations revealed that RHF and MP2 charges are substantially different and electron correlation must be included in the charges calculations of this type of molecules.29
The Lennard-Jones potential parameters were optimised using the all-atom OPLS parameters as a starting trial set.25 They were then adjusted through MC interactive simulations to provide a satisfactory fitting for experimental results of density and enthalpy of vaporisation of acetaldehyde at 18 °C and 1 atm. This adjustment was performed manually. This procedure is preferable over automatic ones, since they often lead to a combination of unphysical parameters that reproduce the input data.9
Monte Carlo simulations
Thermodynamic and structural properties have been computed for the pure liquid by MC simulations.31 Metropolis importance sampling and periodic boundary conditions17 have been used in the NPT ensemble. Cubic boxes containing 500 molecules were simulated at 1 atm and 18 °C. Starting from a molecular distribution in a face centred cell, new configurations were generated by randomly rotating and translating a molecule along randomly chosen Cartesian directions. The ranges for monomer translations and rotations were established to provide an acceptance/trial ratio of about 45% for new configurations. A volume movement was attempted at every 500 configurations and the range for volume change was chosen to provide the same acceptance/trial ratio as above. After a volume movement the intermolecular distances were scaled suitably.
The computational cost of calculating all nonbonded interactions in a system scales as the square of the number of atoms and becomes expensive for a large number of them. It is common practice to use a cut off beyond which these interactions are neglected. In this work, to calculate the intermolecular energy (equation 1), a full intermolecular interaction was considered whenever any site-to-site distance was lower than a cut-off radius rc = 11.0 Å. The contributions of Lennard-Jones interactions beyond the cut-off radius were taken into account using the methodology presented elsewhere.23 In this methodology the long-range contribution from the Lennard-Jones potential is calculated by integrating the potential function from the cut-off radius to infinity, assuming that the pair correlation function is unitary. On the other hand, as it has been widely used in this field of study, long-range corrections to take into account Coulombic interactions beyond cut-off radius were not included in the present work. A brief analysis on the aspects that validate this assumption was done recently.32 When the systems under investigation are constituted of neutral molecules, the main long-range contribution from the electrostatic potential to the cohesive energy comes from the dipole-dipole interaction. This contribution decrease asymptotically with 1/r3, where r is roughly the distance between the centre of mass of two interacting molecules and therefore the dipole-dipole interaction averaged over the angular orientation decrease asymptotically with 1/r6.33 Consequently, the long-range electrostatic contribution to the electrostatic energy is lower than could be expected for a strictly Coulombic system. This is in accord with recent results34 using the generalised Born model.35,36 Also in good agreement with this, it is observed that site-site pair correlation functions, thermodynamic properties and single particle dynamics of polar liquids, appears to be quite insensitive to the long-range forces in the system.21
The calculations started with an equilibration phase with 1.6x107 configurations and the averages were then obtained from a new segment with other 1.6x107 configurations. Statistical uncertainties were calculated from separate averages over blocks of 2x105 configurations.
Results and Discussion
Potential parameters for acetaldehyde
Table 1 lists the final set of potential parameters obtained for acetaldehyde. The parameterisation of a new set of parameters for acetaldehyde is justified because the OPLS charge for aldehydic hydrogen (zero), does not fit properly the weak hydrogen bond features presented for molecules like acetaldehyde.37 The study of these weak hydrogen bonds is the main aim of our work.
It is usual in liquid phase simulations in which molecular dynamic polarisation effects have not been included, to consider as desirable a theoretical molecular dipole moment around 15-20 % greater than the experimental one in the gas phase.28,38,39 This overestimation in the dipole moment has been used as a manner to compensate for neglecting polarisation effects in the simulations. However, the dipole moment for the acetaldehyde molecular charge distribution optimised in this work is µ = 2.6 D, very similar, therefore, to the experimental value µ = 2.7 D.40
Theoretical values of thermodynamic properties for acetaldehyde obtained with OPLS and the present model are shown in Table 2. The experimental values are also reported for comparison.
Radial correlation function and the liquid structure
Radial distribution functions (rdf) play an important role in the understanding of the liquid structure. Experimental information about rdf can be obtained by neutron scattering and X-ray diffraction. Nevertheless, the large number of overlapping distances, both within and between molecules, and the large number of different site-site correlation functions added together in the diffraction pattern, make the experimental investigation of liquid structure difficult.32 Therefore, the use of molecular modeling and computer simulations to acquire the various site-site correlation functions, can be useful in providing improved insight about the liquid structure.
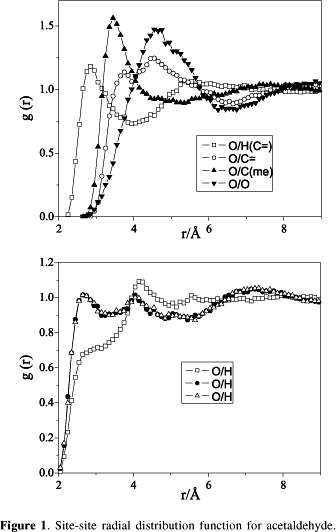
Figure 1 shows the rdf computed with the present model for acetaldehyde. The broadness and positions of the peaks indicate that acetaldehyde is a liquid relatively well structured compared to other organic liquids like tetrahydrofuran and acetone,24,32 which present site-site correlation peaks above 3.0 Å and broad peaks instead of the quite sharp peaks observed in the present case. The first peak in the site-site correlation between O and H(C=), about 2.8 Å, suggests that this correlation can play a significant role in the liquid structure, as has been discussed elsewhere.41-44 The distance O/H(C=) is characteristic of weak hydrogen bonds.45 The same can be addressed for the correlation between oxygen and the methyl hydrogens. As it can be noticed, 2 hydrogens of the methyl group are identically correlated with oxygen while the third is further apart. The shortest O/H distance is about 2.6 Å which permit to classify this interaction also as a weak hydrogen bond, using the same criterion used for the earlier case. The features of weak hydrogen bonds and the criteria used to classify them have been extensively discussed very recently by many authors46-57 and have already been used by us in previous works.12,14 It is important to recall that hydrogen bonds are additive interactions45 and, because of this characteristic, even weak hydrogen bonds can play an important role in stabilising the structure of a material. This can be the reason behind the observation that the methyl carbon is closer to oxygen than the carbonyl carbon. This aspect, however, deserves a closer attention, since other aspects are involved in these interactions. One has to take into account that the methyl carbon is 0.25 Å smaller in diameter than the carbonyl one, and the lower distance verified between oxygen and methyl carbon compared to the distance between oxygen and the carbonyl carbon could be just consequence of this fact. On the other hand, the charge on the methyl carbon is quite negative, as the charge of the oxygen, while the charge of the carbonyl carbon is positive. Considering Coulombic effects, it would be expected an approximation of oxygen to the carbonyl carbon, due to the attraction between them and the repulsion between oxygen and the methyl carbon. However, the contrary behaviour is observed. In this scenario, one could suppose that the attractive interaction between the methyl hydrogens and the oxygen is responsible for this found, but more details to this respect are not possible to be addressed from the present simulations. In this sense, higher level calculations could be interesting to clarify this issue.
The O/C(me) and O/C= pair correlations present some interesting features that is worth being discussed. The O/C= correlation function presents two peaks, one of them is in a position almost overlaping the peak for O/C(me) correlation. This means that the oxygen is roughly localised in the bissecting line of the angle OCC of neighbouring molecules. The second peak about 4.4 Å as well as some shoulders in the O/H(C=) and O/O rdf suggest that the bulk of the liquid should contain more complicated species than just "dimers". Molecular mechanics at MM2 level58 was used in order to minimise a structure that could explain the rdf's shown in Figure 1. After the corresponding calculations it was possible to rise the "trimer" shown61 in Figure 2. The molecules are not coplanar and there is an extraordinary agreement between the geometry of the trimer and the plots of g(r) shown in Figure 1. Little differences in the distances between the same sites taking pairs of molecules (tenth of Å) are found, which explains the pattern observed in the pair correlation peaks. However, the main feature associated with that trimer is that there are two O/C= pairs located at distances that closely agree with the O/C= pair rdf shown in the Figure 1. A standard procedure in this field is the calculation of the site-site coordination number by properly integrating the correspondent correlation function over the first maximum up the position of the first minimum.23 The coordination number  is given for
is given for  , where r is the liquid density and r is the integrating radius. r0 is the radius at the first minimum.
, where r is the liquid density and r is the integrating radius. r0 is the radius at the first minimum.
The number of hydrogen bonds O/H(C=) obtained was 1.8. This result corroborates that a trimer is the representative structure in the liquid bulk. Thus, each one of the oxygens in the trimer complex is near to two equidistant carbonilic hydrogens. The C/H(C=) distance calculated in the complex also compares well with the corresponding site-site distance in the rdf graphic. These results indicate that the weak hydrogen bonds between the carbonylic oxygen and hydrogen play a important role in the stabilisation of the liquid structure, what agrees with a great number of other results previously published.41-57 Besides, the relative position of one molecule with respect to an other, due to the correlation between the oxygen and the methyl hydrogens, could be interpreted as more a indication of the weight of the weak hydrogen bonds in the structure. Taking this result into account one can suppose that this type of interaction can play an important role in the structure of molecules like proteins for example, mainly if normal hydrogen bonds are not involved.
The energy of the acetaldehyde trimer was also investigated using molecular mechanics at MM2 level,58 as it was done in a previous work for acetone.32 The acetaldehyde trimer here reported has a stabilisation energy at MM2 level of 10.23 kcal/mol, whilst the stabilisation energy of the acetone dimer32 is 2.91 kcal/mol. Thus the trimer of acetaldehyde seems to be particularly more stable than the dimer of acetone. The O/H interactions can be playing a considerable role in this stabilisation. The high energy of stabilisation of the acetaldehyde trimer permits to understand why the liquid is so well structured compared to acetone, according to the rdf's of both of them.
Finally, the rdf obtained with the acetaldehyde molecule parameterised in this work are quite similar to those calculated with the OPLS model despite the huge difference in the atomic charges of the two models. The similarity of their dipole moment, however, would indicate that the dipole moment would play an important role in the liquid structure, as have already been pointed out previously.59 According to Amey,59 there is a preferential inter-molecular geometry arising from dipole-dipole orientation, and any form of molecular array in the liquid must be related to the dipole moment. On the other hand, Luzar et al.60 have shown that a substantial part of the site-site correlation in DMSO is due to packing effects and, hence, cannot be entirely credited to molecular association. Taking into account these results it is difficult to quantify how much the liquid is structured because of the dipole-dipole/packing effects and how much because of atomic interactions. A theoretical investigation aiming to quantify these effects would be extremely important. Of course, even if the molecules were relatively oriented because of the dipolar interaction, specific atomic roles would complete the scenario giving the final structure of the liquid.
Conclusions
A new model for acetaldehyde has been optimised, and some thermodynamic and structural features of it have been calculated using Monte Carlo simulations. The site charges used, obtained from Mulliken distribution at MP2 level, seem to provide a good charge distribution for the molecule. The charge distribution fits well the experimental value the dipole moment. The model here reported has a suitable ability to mimic the energetic and structural properties of liquid acetaldehyde that are related to density and heat of vaporisation. The rdf for site-site correlations present quite sharp peaks indicating that acetaldehyde is a well organised liquid. Molecular mechanics calculations showed that the predominant species in the bulk is a trimer, whose structure was here reported. This trimer seems to be quite stable. The relative positions among atoms in this complex suggest the formation of O/carbonyl hydrogen and O/methyl hydrogens weak hydrogen bonds. These interactions play an important role in the stabilisation of the liquid structure.
Acknowledgments
J.M.M.C. is grateful to FAPESP Fundação de Amparo à Pesquisa do Estado de São Paulo and FUNDUNESP Fundação para o Desenvolvimento da Unesp, for financial support.
References
1. Caccamo, C.; Hansen, J-P.; Stell, G.; New Approaches to Problems in Liquid State Theory; NATO ASI series, Series C: Mathematical and Physics Sciences, Kluwer Academic: Dordrecht, 1999, p. 529.
2. Kessler, Y.M.; Puhovski, Y.P.; Kiselev, M.G.; Vaisman, I.I. In Chemistry of Non Aqueous Solutions: Current Progress; Mamantov, G.; Popov, A.I., eds.; VCH: New York, 1994, p. 307.
3. Teixeira-Dias, J.J., ed.; Molecular Liquids: New Perspectives in Physics and Chemistry, NATO ASI Series, Kluwer Academic: Dordrecht, 1991.
4. Rappaport, D.C.; The Art of Molecular Dynamics Simulation, Cambridge University Press: Cambridge, 1995.
5. Allen, M.; Tildesley, D.J., eds.; Computer Simulations in Chemical Physics, Kluwer Academic Press: Dordrecht, 1993.
6. Halgreen, T. A.; J. Am. Chem. Soc. 1992, 114, 7827.
7. Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J.; J. Am. Chem. Soc. 1996, 118, 11225.
8. Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A.; J. Am. Chem. Soc. 1995, 117, 5179.
9. MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; Joseph-McCarthy, D.; Kuchnir, L.; Kuczera, K.; Lau, F.T.K.; Mattos, C.; Michnick, S.; Ngo, T.; Nguyen, D.T.; Prodhom, B.; Reiher, W.E.; Roux, B.; Schlenkrich, M.; Smith, J.C.; Stote, R.; Straub, J.; Watanabe, M.; Wiorkiewicz-Kuczera, J.; Yin, D.; Karplus, M.; J. Phys. Chem. B. 1998, 102, 3586.
10. Ben Naim, A.; Water and Aqueous Solution, Plenum Press: New York, 1986.
11. Hansen, J.; McDonald, I.R.; Theory of Simple Liquids, Academic Press: London, 1986.
12. Cordeiro, J.M.M.; Int. J. Quantum Chem. 1997, 65, 709.
13. Cordeiro, J.M.M.; Mol. Eng. 1999, 8, 303.
14. Cordeiro, J.M.M.; Freitas, L.C.G.; Z. Naturforsch 1999, 54a, 110.
15. Chen, L.P.; Gross, T.; Ludemann, H.D.; Z. Phys. Chem., part 2 2000, 214, 239.
16. Parker, A.J.; Chem. Rev. 1969, 69, 1; Abraham, M.H. Prog. Phys. Org. Chem. 1974, 11, 185.
17. Metropolis, N.A.; Rosenbluth, W.; Rosenbluth M.N.; Teller, A.H.; Teller, E.; J.Chem. Phys. 1953, 21, 108.
18. Alder, B.J.; Wainwright, T.E.; J.Chem. Phys. 1959, 31, 459; ibid 1960, 33, 1439.
19. Strastsma, T.P.; McCammon, J.A.; Annu. Rev. Phys. Chem. 1992, 43, 407.
20. Davidson, E. R.; Chem. Rev. 1993, 93, 2337; Dykstra, C.E.; ibid, 2339; Berry, R.S.; ibid, 2379.
21. Ladanyi, B.M.; Skaf, M.S.; Ann. Rev. Phys. Chem. 1993, 44, 335.
22. Rappé, A.K.; Casewit, C.J.; Molecular Mechanics Across Chemistry, University Science Books: Suasalito, 1997.
23. Allen, M.; Tildesley, D. J.; Computer Simulation of Liquids, Oxford University Press: Oxford, 1987.
24. Freitas, L.C.G.; Cordeiro, J.M.M.; J. Mol. Struct. (Theochem) 1995, 333, 189.
25. Jorgensen, W.L., personal communication.
26. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Grill, P.M.W.; Johnson, B.G.; Robb, M.A.; Cheeseman, J.R.; Keith, T.A.; Petersson, G.A.; Montgomery, J.A.; Raghavachari, K.; Al-Laham, M.A.; Zakrzewski, V.G.; Ortiz, J.V.; Foresman, J.B.; Cioslowski, J.; Stefanov, B.B.; Nanayakkara, A.; Challacombe, M.; Peng, C.Y.; Ayala, P.Y.; Chen, W.; Wong, M.W.; Andres, J.L.; Replogle, E.S.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Binkley, J.S.; Defrees, D.J.; Baker, J.; Stewart, J.P.; Head-Gordon, M.; Gonzalez, C.; Pople, J.A.; GAUSSIAN 94, Gaussian, Inc.: Pittsburgh PA, 1995.
27. Breneman, M.; Wiberg, K.B.J.; J. Comp. Chem. 1990, 11, 361.
28. Carlson, H.A.; Nguyen, T.B.; Orosco, M.; Jorgensen, W.L.; J. Comp. Chem. 1993, 14, 1240.
29. Cordeiro,J.M.M.; unpublished results, 2000.
30. Merz Jr., K.M.; J. Comp. Chem. 1992, 13, 749; Luque, F.J.; Orosco, M.; Illas, F.; Rubio, J.; J. Am. Chem. Soc. 1991, 113, 5203.
31. Gomide Freitas, L.C.; DIADORIM - Program for Monte Carlo Simulations of Líquids, Universidade Federal de São Carlos: São Paulo, Brazil, 1993.
32. Freitas, L.C.G.; Cordeiro, J.M.M.; Garbujo, F.L.L.; J. Mol. Liq. 1999, 79, 1.
33. Israelachvili, J.N.; Intermolecular and Surface Forces, Academic Press: London, 1985.
34. Belletato, P.; Freitas, L.C.G.; Elizabeth, P.G.A.; Santos, P.S.; Phys. Chem. Chem. Phys. 1999, 1, 4769.
35. Newmann, M.J.; Chem. Phys. 1985, 82, 5663.
36. Alper, H.E.; Levy, R.M.; J. Chem. Phys. 1989, 91, 1242.
37. Steiner, T.; Chem. Commun. 1997, 72.
38. Dykstra, C.E.; Chem. Rev. 1993, 93, 2339.
39. Jorgensen, W.L.; Chemtracts: Org. Chem. 1991, 4, 91.
40. Lide, D.R., ed.-in-chief; Handbook of Chemistry and Physics, 76th ed., CRC Press Inc: USA, 1995.
41. Steiner, T.; Chem. Commun. 1999, 313.
42. Steiner, T.; Desiraju, G.R.; Chem. Commun. 1998, 891.
43. Desiraju, G.R.; Acc. Chem. Res. 1991, 24, 290; ibid 1996, 29, 441.
44. Desiraju, G.R.; Angew. Chem. Int. Ed. Eng. 1995, 34, 2311.
45. Desiraju, G.R.; Steiner, T.; The Weak Hydrogen Bond, Oxford University Press: Oxford, 1999.
46. Steiner, T.; Lutz, B.; Van der Maas, J.; Schreurs, A.M.M.; Kroon, J.; Tamm, M.; Chem. Commun. 1998, 171.
47. Steiner, T.; Chem. Commun. 1997, 727.
48. Steiner, T.; Saenger, J.; J. Chem. Soc., Chem. Commun. 1995, 2087; Braga, D.; Grepioni, F.; Byrne, J.J.; Wolf, A.; ibid 1995, 125; Behrens, P.; Van de Goor, G.; Freyhardt, C.C.; Angew. Chem., Int. Ed. Engl. 1995, 34, 2895.
49. Derewenda, Z.; Derewenda, S.U.; Kobos, P.M.; J. Mol. Biol. 1994, 241, 83; Derewenda, Z.S.; Lee, L.; Derewenda, U.; ibid 1995, 252, 248; Leonard, G.A.; McAuley-Hecht, K.; Brown, T.; Hunter, W.N.; Acta Cryst. 1995, D51, 136; Wahl, M.C.; Rao, S.T.; Sundaralingam, M.; Nature Struct. Biol. 1996, 3, 24.
50. Steiner, T.; J. Chem. Soc. Chem. Commun. 1994, 2341.
51. Jeffrey, G.A.; Saenger, W.; Hydrogen Bonding in Biological Structures, Springer: Berlim, 1991.
52. Turi, L.; Dannenberg, J.J.; J. Phys. Chem. 1993, 97, 7899; Novoa, J.J.; Tarron, B.; Whangbo, M.H.; Williams, J.M.; J. Chem. Phys. 1991, 95, 5179; van Mourik, T.; van Duijneveldt, F.B.; J. Mol. Struct. (Theochem) 1995, 341, 63.
53. Lutz, B.; van der Maas, J.; Kanters, J.A.; J. Mol. Struct. 1994, 325, 203; Steiner, T.; J. Chem. Soc., Perkin Trans. 1995, 2, 1315; Allen, F.H.; Howard, J.A.K.; Hoy, V.J.; Desiraju, G.R.; Reddy, D.S.; Wilson, C.C.; J. Am. Chem. Soc. 1996, 118, 4081.
54. Steiner, T.; Kanters, J.A.; Kroon, J.; Chem. Commun. 1996, 1277.
55. Sharma, C.V.; Desiraju, G.R.; J. Chem. Soc., Perkin Trans. 1994, 2, 2345.
56. Steiner, T.; Saenger, W.; J. Am. Chem. Soc. 1993, 115, 4540; Steiner, T.; Saenger, W.; Acta Cryst. 1993, D49, 592; Steiner, T.; Acta Cryst. 1995, D51, 93.
57. Steiner, T.; Koellner, G.; Gessler, K.; Saenger, W.; J. Chem. Soc. Chem. Commun. 1995, 511.
58. PCMODEL, Serena Software, Bloomington, USA, 1989.
59. Amey, R.L.; J. Phys. Chem. 1968, 72, 3358.
60. Luzar, A.; Soper, A.K.; Chandler, D.; J. Chem. Phys. 1993, 99, 6836.
61. Sayle, R.; RASMOL V2.5, Molecular Visualisation Program; Glaxo Research and Development: Greenford, Middlesex, U.K, 1995.
Received: July 17, 2002
Published on the web: April 20, 2004
FAPESP helped in meeting the publication costs of this article
- 1. Caccamo, C.; Hansen, J-P.; Stell, G.; New Approaches to Problems in Liquid State Theory; NATO ASI series, Series C: Mathematical and Physics Sciences, Kluwer Academic: Dordrecht, 1999, p. 529.
- 2. Kessler, Y.M.; Puhovski, Y.P.; Kiselev, M.G.; Vaisman, I.I. In Chemistry of Non Aqueous Solutions: Current Progress; Mamantov, G.; Popov, A.I., eds.; VCH: New York, 1994, p. 307.
- 3. Teixeira-Dias, J.J., ed.; Molecular Liquids: New Perspectives in Physics and Chemistry, NATO ASI Series, Kluwer Academic: Dordrecht, 1991.
- 4. Rappaport, D.C.; The Art of Molecular Dynamics Simulation, Cambridge University Press: Cambridge, 1995.
- 5. Allen, M.; Tildesley, D.J., eds.; Computer Simulations in Chemical Physics, Kluwer Academic Press: Dordrecht, 1993.
- 6. Halgreen, T. A.; J. Am. Chem. Soc 1992, 114, 7827.
- 7. Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J.; J. Am. Chem. Soc 1996, 118, 11225.
- 8. Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A.; J. Am. Chem. Soc 1995, 117, 5179.
- 9. MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; Joseph-McCarthy, D.; Kuchnir, L.; Kuczera, K.; Lau, F.T.K.; Mattos, C.; Michnick, S.; Ngo, T.; Nguyen, D.T.; Prodhom, B.; Reiher, W.E.; Roux, B.; Schlenkrich, M.; Smith, J.C.; Stote, R.; Straub, J.; Watanabe, M.; Wiorkiewicz-Kuczera, J.; Yin, D.; Karplus, M.; J. Phys. Chem. B 1998, 102, 3586.
- 10. Ben Naim, A.; Water and Aqueous Solution, Plenum Press: New York, 1986.
- 11. Hansen, J.; McDonald, I.R.; Theory of Simple Liquids, Academic Press: London, 1986.
- 12. Cordeiro, J.M.M.; Int. J. Quantum Chem 1997, 65, 709.
- 13. Cordeiro, J.M.M.; Mol. Eng 1999, 8, 303.
- 14. Cordeiro, J.M.M.; Freitas, L.C.G.; Z. Naturforsch 1999, 54a, 110.
- 15. Chen, L.P.; Gross, T.; Ludemann, H.D.; Z. Phys. Chem, part 2 2000, 214, 239.
- 16. Parker, A.J.; Chem. Rev 1969, 69, 1;
- Abraham, M.H. Prog. Phys. Org. Chem 1974, 11, 185.
- 17. Metropolis, N.A.; Rosenbluth, W.; Rosenbluth M.N.; Teller, A.H.; Teller, E.; J.Chem. Phys 1953, 21, 108.
- 18. Alder, B.J.; Wainwright, T.E.; J.Chem. Phys 1959, 31, 459;
- 19. Strastsma, T.P.; McCammon, J.A.; Annu. Rev. Phys. Chem 1992, 43, 407.
- 20. Davidson, E. R.; Chem. Rev 1993, 93, 2337;
- Dykstra, C.E.; ibid, 2339;
- Berry, R.S.; ibid, 2379.
- 21. Ladanyi, B.M.; Skaf, M.S.; Ann. Rev. Phys. Chem 1993, 44, 335.
- 22. Rappé, A.K.; Casewit, C.J.; Molecular Mechanics Across Chemistry, University Science Books: Suasalito, 1997.
- 23. Allen, M.; Tildesley, D. J.; Computer Simulation of Liquids, Oxford University Press: Oxford, 1987.
- 24. Freitas, L.C.G.; Cordeiro, J.M.M.; J. Mol. Struct. (Theochem) 1995, 333, 189.
- 25. Jorgensen, W.L., personal communication.
- 26. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Grill, P.M.W.; Johnson, B.G.; Robb, M.A.; Cheeseman, J.R.; Keith, T.A.; Petersson, G.A.; Montgomery, J.A.; Raghavachari, K.; Al-Laham, M.A.; Zakrzewski, V.G.; Ortiz, J.V.; Foresman, J.B.; Cioslowski, J.; Stefanov, B.B.; Nanayakkara, A.; Challacombe, M.; Peng, C.Y.; Ayala, P.Y.; Chen, W.; Wong, M.W.; Andres, J.L.; Replogle, E.S.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Binkley, J.S.; Defrees, D.J.; Baker, J.; Stewart, J.P.; Head-Gordon, M.; Gonzalez, C.; Pople, J.A.; GAUSSIAN 94, Gaussian, Inc.: Pittsburgh PA, 1995.
- 27. Breneman, M.; Wiberg, K.B.J.; J. Comp. Chem 1990, 11, 361.
- 28. Carlson, H.A.; Nguyen, T.B.; Orosco, M.; Jorgensen, W.L.; J. Comp. Chem 1993, 14, 1240.
- 29. Cordeiro,J.M.M.; unpublished results, 2000.
- 30. Merz Jr., K.M.; J. Comp. Chem 1992, 13, 749;
- Luque, F.J.; Orosco, M.; Illas, F.; Rubio, J.; J. Am. Chem. Soc 1991, 113, 5203.
- 31. Gomide Freitas, L.C.; DIADORIM - Program for Monte Carlo Simulations of Líquids, Universidade Federal de São Carlos: São Paulo, Brazil, 1993.
- 32. Freitas, L.C.G.; Cordeiro, J.M.M.; Garbujo, F.L.L.; J. Mol. Liq 1999, 79, 1.
- 33. Israelachvili, J.N.; Intermolecular and Surface Forces, Academic Press: London, 1985.
- 34. Belletato, P.; Freitas, L.C.G.; Elizabeth, P.G.A.; Santos, P.S.; Phys. Chem. Chem. Phys 1999, 1, 4769.
- 35. Newmann, M.J.; Chem. Phys 1985, 82, 5663.
- 36. Alper, H.E.; Levy, R.M.; J. Chem. Phys 1989, 91, 1242.
- 37. Steiner, T.; Chem. Commun 1997, 72.
- 38. Dykstra, C.E.; Chem. Rev 1993, 93, 2339.
- 39. Jorgensen, W.L.; Chemtracts: Org. Chem 1991, 4, 91.
- 40. Lide, D.R., ed.-in-chief; Handbook of Chemistry and Physics, 76th ed., CRC Press Inc: USA, 1995.
- 41. Steiner, T.; Chem. Commun 1999, 313.
- 42. Steiner, T.; Desiraju, G.R.; Chem. Commun 1998, 891.
- 43. Desiraju, G.R.; Acc. Chem. Res 1991, 24, 290;
- 44. Desiraju, G.R.; Angew. Chem. Int. Ed. Eng 1995, 34, 2311.
- 45. Desiraju, G.R.; Steiner, T.; The Weak Hydrogen Bond, Oxford University Press: Oxford, 1999.
- 46. Steiner, T.; Lutz, B.; Van der Maas, J.; Schreurs, A.M.M.; Kroon, J.; Tamm, M.; Chem. Commun 1998, 171.
- 47. Steiner, T.; Chem. Commun 1997, 727.
- 48. Steiner, T.; Saenger, J.; J. Chem. Soc., Chem. Commun 1995, 2087;
- Braga, D.; Grepioni, F.; Byrne, J.J.; Wolf, A.; ibid 1995, 125;
- Behrens, P.; Van de Goor, G.; Freyhardt, C.C.; Angew. Chem., Int. Ed. Engl 1995, 34, 2895.
- 49. Derewenda, Z.; Derewenda, S.U.; Kobos, P.M.; J. Mol. Biol 1994, 241, 83;
- Derewenda, Z.S.; Lee, L.; Derewenda, U.; ibid 1995, 252, 248;
- Leonard, G.A.; McAuley-Hecht, K.; Brown, T.; Hunter, W.N.; Acta Cryst 1995, D51, 136;
- Wahl, M.C.; Rao, S.T.; Sundaralingam, M.; Nature Struct. Biol 1996, 3, 24.
- 50. Steiner, T.; J. Chem. Soc. Chem. Commun 1994, 2341.
- 51. Jeffrey, G.A.; Saenger, W.; Hydrogen Bonding in Biological Structures, Springer: Berlim, 1991.
- 52. Turi, L.; Dannenberg, J.J.; J. Phys. Chem 1993, 97, 7899;
- Novoa, J.J.; Tarron, B.; Whangbo, M.H.; Williams, J.M.; J. Chem. Phys 1991, 95, 5179;
- van Mourik, T.; van Duijneveldt, F.B.; J. Mol. Struct. (Theochem) 1995, 341, 63.
- 53. Lutz, B.; van der Maas, J.; Kanters, J.A.; J. Mol. Struct 1994, 325, 203;
- Steiner, T.; J. Chem. Soc., Perkin Trans 1995, 2, 1315;
- Allen, F.H.; Howard, J.A.K.; Hoy, V.J.; Desiraju, G.R.; Reddy, D.S.; Wilson, C.C.; J. Am. Chem. Soc 1996, 118, 4081.
- 54. Steiner, T.; Kanters, J.A.; Kroon, J.; Chem. Commun 1996, 1277.
- 55. Sharma, C.V.; Desiraju, G.R.; J. Chem. Soc., Perkin Trans 1994, 2, 2345.
- 56. Steiner, T.; Saenger, W.; J. Am. Chem. Soc 1993, 115, 4540;
- Steiner, T.; Saenger, W.; Acta Cryst. 1993, D49, 592;
- Steiner, T.; Acta Cryst 1995, D51, 93.
- 57. Steiner, T.; Koellner, G.; Gessler, K.; Saenger, W.; J. Chem. Soc. Chem. Commun 1995, 511.
- 58. PCMODEL, Serena Software, Bloomington, USA, 1989.
- 59. Amey, R.L.; J. Phys. Chem 1968, 72, 3358.
- 60. Luzar, A.; Soper, A.K.; Chandler, D.; J. Chem. Phys 1993, 99, 6836.
- 61. Sayle, R.; RASMOL V2.5, Molecular Visualisation Program; Glaxo Research and Development: Greenford, Middlesex, U.K, 1995.
Publication Dates
-
Publication in this collection
16 June 2004 -
Date of issue
June 2004
History
-
Received
17 July 2002 -
Accepted
20 Apr 2004