Abstracts
The NiS2 and Fe2O3 active phases supported on medium surface area beta-silicon carbide (SiC) showed high activity, selectivity and stability in the direct oxidation of H2S into elemental sulfur. The catalysts were tested under a large range of temperatures, from room temperature using Ni (trickle-bed reactor) to over-dewpoint temperatures using Fe (fixed bed reactor). For both cases, the formation on stream of a Ni and Fe oxysulfide high active phase was proposed by oxidation of NiS2 and sulfidation of Fe2O3, respectively. The absence of microporosity in the support contributed to the high selectivity into sulfur. At low temperatures (below 100 ºC), the high stability of beta-SiC supported catalysts towards the solid sulfur loading was explained by a specific mode of sulfur deposition, involving the role of water in the feed and the heterogeneous nature of the SiC surface, being partly hydrophilic and hydrophobic.
silicon carbide; catalyst support; sulfur recovery; H2S oxidation; hydrophilicity; hydrophobicity
As fases ativas NiS2 e Fe2O3 suportadas em beta-carbeto de silício (SiC) com média área específica mostraram alta atividade, seletividade e estabilidade na oxidação direta do H2S a enxofre elementar. Os catalisadores foram testados a temperaturas que variaram da temperatura ambiente, no caso do Ni em reator de leito gotejante, até temperaturas superiores à do ponto de orvalho do enxofre, no caso do Fe em reator de leito fixo. Para ambos os casos, foi proposta a formação de uma fase bastante ativa de oxisulfeto de Ni ou de Fe, formada pela oxidação do NiS2 e pela sulfuração do Fe2O3. A ausência de microporosidade no suporte contribuiu à alta seletividade do catalisador. A grande estabilidade ao carregamento de enxofre sólido, apresentada pelos catalisadores suportados em SiC em temperaturas inferiores a 100 ºC, foi explicada pela maneira especial da deposição do enxofre, a qual depende do papel da água presente na reação e do caráter heterogêneo (hidrofílico e hidrofóbico) da superfície do suporte.
ARTICLE
New catalysts based on silicon carbide support for improvements in the sulfur recovery. Silicon carbide as support for the selective H2S oxidation
Nicolas Keller; Ricardo Vieira* * e-mail: vieirar@ecpm.u-strasbg.fr ; Jean-Mario Nhut; Cuong Pham-Huu; Marc J. Ledoux
Laboratoire des Matériaux, Surface et Procédés pour la Catalyse, Université Louis Pasteur, UMR 7515 CNRS, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France
ABSTRACT
The NiS2 and Fe2O3 active phases supported on medium surface area b-silicon carbide (SiC) showed high activity, selectivity and stability in the direct oxidation of H2S into elemental sulfur. The catalysts were tested under a large range of temperatures, from room temperature using Ni (trickle-bed reactor) to over-dewpoint temperatures using Fe (fixed bed reactor). For both cases, the formation on stream of a Ni and Fe oxysulfide high active phase was proposed by oxidation of NiS2 and sulfidation of Fe2O3, respectively. The absence of microporosity in the support contributed to the high selectivity into sulfur. At low temperatures (below 100 ºC), the high stability of b-SiC supported catalysts towards the solid sulfur loading was explained by a specific mode of sulfur deposition, involving the role of water in the feed and the heterogeneous nature of the SiC surface, being partly hydrophilic and hydrophobic.
Keywords: silicon carbide, catalyst support, sulfur recovery, H2S oxidation, hydrophilicity, hydrophobicity
RESUMO
As fases ativas NiS2 e Fe2O3 suportadas em b-carbeto de silício (SiC) com média área específica mostraram alta atividade, seletividade e estabilidade na oxidação direta do H2S a enxofre elementar. Os catalisadores foram testados a temperaturas que variaram da temperatura ambiente, no caso do Ni em reator de leito gotejante, até temperaturas superiores à do ponto de orvalho do enxofre, no caso do Fe em reator de leito fixo. Para ambos os casos, foi proposta a formação de uma fase bastante ativa de oxisulfeto de Ni ou de Fe, formada pela oxidação do NiS2 e pela sulfuração do Fe2O3. A ausência de microporosidade no suporte contribuiu à alta seletividade do catalisador. A grande estabilidade ao carregamento de enxofre sólido, apresentada pelos catalisadores suportados em SiC em temperaturas inferiores a 100 ºC, foi explicada pela maneira especial da deposição do enxofre, a qual depende do papel da água presente na reação e do caráter heterogêneo (hidrofílico e hidrofóbico) da superfície do suporte.
Introduction
The removal of hydrogen sulfide (H2S) from the acid gases generated by oil refineries or natural gas plants is a crucial aspect of air pollution control, due to ever increasing standards of efficiency required by the environmental protection pressure. The general trend is to selectively transform H2S into elemental sulfur and steam, by the well-known modified Claus process.1 (equation 1)
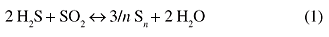
According to the different running processes, typical sulfur efficiencies are only 90-96 % for a two stage reactor unit and 95-98 % for a three stage process, due to the thermodynamic limitations of the Claus reaction.2 This means that for a conventional sulfur plant, the SO2 emissions may amount to many thousand tons per year released into the atmosphere, and a large amount of sulfur compounds remains in the tail gas, i.e., SO2 (» 6000 ppm), H2S (» 12000 ppm), COS and CS2 (» 700 ppm). New catalysts and processes for the tail-gas treatment of the industrial Claus plants were considerably developed to reduce the last remaining sulfur and to meet the strict legislation requirements for the sulfur release into the atmosphere. These processes are based on the direct oxidation by oxygen of remaining traces of H2S, or on the H2S absorption/recycling technologies which require expensive investments and running costs.3
Up to now, two main catalytic processes dealing with the selective oxidation of H2S by oxygen into elemental sulfur have been developed. The Sulfreen-like processes (Elf-Lurgi) based on Cu or Ni catalysts supported on modified alumina work below the sulfur dewpoint.4 In such conditions, the reaction is performed in a discontinuous mode: the solid sulfur formed is continuously deposited onto the catalyst surface and periodical regeneration has to be performed to remove the solid sulfur from the catalyst surface. In order to avoid such regenerative treatments required for sub-dewpoint processes, the reaction can be performed in over-dewpoint conditions, typically at temperatures greater than 180 ºC. The high temperature Superclaus process, developed by Comprimo, Gastec and the University of Utrecht (The Netherlands), working in a continuous mode above the sulfur dewpoint (> 180 ºC), is based on Fe and Fe/Cr catalysts supported on alumina or silica.5 There are three major critical drawbacks for the usually used catalysts in discontinuous and continuous processes. First of all, the selectivity into sulfur can be decreased by the direct oxidation of H2S into SO2, and by the successive oxidations of H2S into sulfur and then into SO2. In addition, the presence of water (up to 30 vol. % in the Claus tail-gas) can lead to a loss in sulfur yield, by formation both of H2S and SO2via a so-called retro-Claus reaction. The poor stability of most of the used oxide supports is also a critical drawback, as they are very sensitive to the problem of sulfation during the reaction in the presence of steam, sulfur, SO2 and oxygen. Most of the used oxides reacts with sulfur-containing compounds under such conditions, leading with time on stream to a decrease in activity or even to the destruction of the catalyst, mainly by encapsulation of the active phase by the sulfated support.6 Moreover, from an economic point of view, such processes require a high capital investment and running costs.4,5
Parallely to its development for use in a broad range of applications including biomedical materials, high temperature semiconducting devices, optical elements and lifeweight/high strength structures, silicon carbide (SiC) has been receiving a growing interest as a support material in the catalysis field.7-10 Properties required for use as catalyst support, such as the high thermal conductivity, high resistance towards oxidation, high mechanical strength and chemical inertness, are shown by SiC. 11,12 The generally used traditional supports such as the high surface area activated carbon, alumina or other oxides, display several restricting drawbacks, which are usually detrimental for several catalytic applications: (i) a low mechanical stability, leading to surface area collapse or even to the catalyst destruction, (ii) a low thermal stability, which leads to modifications of the support and also induces the formation of hot spots during the reaction or the regeneration phases, (iii) the chemical reactivity of the support with the reaction mixture or the active phase itself, leads to the formation of a new compound, resulting in loss of the active phase and as a consequence, the catalytic reactivity.13 In addition, the strong interaction of the alumina support with the active phase also hinders recovery of the latter at the end of the catalyst life-time. Due to its interesting physico-chemical properties, SiC could thus overcome the restricting drawbacks of the generally used traditional supports, and also be a promising candidate to replace conventional industrial supports for several catalytic reactions. Several reviews recently summarized the physico-chemical properties of SiC for use as catalyst support compared to the detrimental drawbacks of traditional supports.11,12 However, SiC must be prepared in a medium or high surface area range (20-200 m2 g-1) in order to be useful as a catalyst support, and its development has been limited by the inability to obtain it. The main drawback of SiC, i.e., the very low surface area of the commercially available material, was overcome by new preparation methods, allowing higher surface area carbides with high synthesis yield. Another drawback of the SiC material resides in the difficulty in its shaping since the industrial SiC is in a powder form and is also not directly suitable for use as catalyst support.
The aim of this work is to report the selective oxidation of H2S by oxygen into elemental sulfur, using silicon carbide (SiC) as catalyst support, prepared following a synthesis method called the "Shape Memory Synthesis" (SMS). This synthesis was developed few years ago by Ledoux et al.14 and overcome the disadvantages cited above. Since its discovery, SiC prepared according to the SMS method was efficiently used for several reactions such as hydrodesulfurization, automotive exhaust-pipe reactions, isomerization of linear saturated hydrocarbons, n-butane dehydrogenation-isomerization reactions, reported in detailed reviews published by Ledoux et al.11,15 Due to its interesting physico-chemical properties, SiC seems to be a promising substitute to traditional oxidic supports for the selective oxidation of H2S by oxygen into elemental sulfur.
Experimental
Catalyst preparation
The laboratory scale of the SMS method consisted in the gas-solid reaction between SiO vapors and pre-shaped activated charcoal under dynamic vacuum at a temperature around 1200-1300 ºC following the reaction equation (2), the SiO vapors being first generated by reaction between silicon and silica at lower temperatures (3):
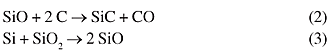
The macrostructural features of the starting carbon determined the resulting SiC morphology (e.g. grains, extrudates, spheres, monoliths) leading to the shape memory concept. The SMS method also allowed the preparation of SiC with controllable and specific size and shape, by controlling the carbon based precursor. Such a control is a real advantage compared to the traditional SiC, for which binders are required to obtain the final macroscopic shape. The preparation of SiC in a grain form (0.4 1 mm grain size) used in the present work has been achieved by the Pechiney Company at Voreppe (France).16 This industrial method is an extension of the laboratory scale of the SMS method and consisted in the pre-mixing of a carbon powder with a fine powder of silicon into a polyfuran resin used as oxygen source. This was further pre-shaped, dried and heated in a rotative oven at about 1300 ºC in a counter flow of argon gas. Such a process allowed the preparation of silicon carbide in a continuous mode without the use of vacuum media. A new company called SICAT17 has been set up in order to industrially produce silicon carbide suitable for use as catalyst support for customers.
The nickel-based catalysts were prepared by incipient wetness impregnation of the SiC support with an aqueous solution of Ni(NO3)2.H2O (Merck). They were dried at 120 ºC for 14 h and then calcined at 350 ºC for 2 h in order to decompose the nitrate salt and to form nickel oxide. NiS2/SiC was obtained by sulfidation of NiO/SiC by the reaction with a H2S (4 vol. %)/He flow at 300 ºC for 4 h (100 mL min-1 for 5 mL of catalyst, corresponding to a catalyst weight of 3.35 g). The NiS2 nature of the sulfided supported phase was proved by X-ray diffraction (Figure 1).The same impregnation technique was used for the iron-based active phase, using an aqueous solution of Fe(NO3)3.9H2O (Merck). The final calcination was in this case performed at 400 ºC to form the Fe2O3 oxide. The loading of Ni or Fe, expressed as metal, was set at 5 wt. % and confirmed by the atomic absorption technique, performed at the Service Central d'Analyse of the CNRS (Vernaison, France).
Micropilot description
The H2S oxidation was performed in a Pyrex fixed-bed reactor. The flow rate of gases (O2 and H2S) was monitored by mass flowmeters (Tylan FC280A with a Tylan RC280 control unit), whereas steam was provided by a saturator kept at the required temperature allowing variations of the partial pressure of the water from 0 to 30 vol. %. The analysis of the inlet and outlet gases was performed on-line using a Varian CX-3400 gas chromatograph equipped with a Chrompack JSQ capillary column able to separate O2, H2S, H2O and SO2, a catharometer detector and a calibrated six port loop. The gas hourly space velocity (GHSV) and the weight hourly space velocity (WHSV) were taken respectively as the ratio of the total inlet flow per hour to the volume of catalyst used, and the ratio of the inlet H2S weight per hour to the weight of catalyst used.
Characterization techniques
Structural characterization of the samples was done by powder X-ray diffraction (XRD) measurements, carried out with a Siemens Diffractometer Model D-5000, using a CuKa radiation source. The nature of the crystalline phases present was checked using the database of the Joint Committee on Powder Diffraction Standards (JCPDS).
Surface areas were measured by means of a commercial BET unit (Coulter Model SA 3100) using N2 adsorption at 196 ºC. The surface area was the surface calculated from the N2 isotherm using the BET method. The micropore content was obtained from the t-plot method in conjunction with the usual Harkins-Jura thickness equation.
Scanning Electron Microscopy (SEM) was performed on a JEOL JMS840 working at 20 mA with an accelerating voltage of 20 kV and Transmission Electron Microscopy (TEM) was carried out on a Topcon EM-002 UHR operating at 200 kV with a point to point resolution of 0.17 nm.
Results and Discussion
Medium surface area b-silicon carbide
The bulk chemical nature of the synthesized material was confirmed by the X-ray diffraction pattern, which only exhibited the diffraction lines corresponding to the b-SiC phase, crystallized in a face-centered cubic structure (not shown). Other compounds such as silicon or silica were not detected by XRD, meaning that such compounds, if present, were only in an amorphous form or in very small amounts. The SiC had a specific surface area of 25 m2 g-1, measured according to the BET method. It had a meso- and macroporosity, with no microporous network.15Figures 2A and 2B show respectively the macroporosity of the b-SiC support and the presence of a 1-3 nm thick amorphous oxygen containing surface layer. Previous characterizations by XPS/mapping-Auger coupled to TEM analyses evidenced that the surface of SiC prepared according to the gas-solid SMS was heterogeneous in nature:15,18,19 a fraction of the surface is composed of this amorphous surface phase, made of a mixture of silicon oxycarbide and silica phases, expected to present a hydrophilic character due to the presence of oxygen atoms on the surface, whereas the remaining surface is pure SiC, oxygen-free, expected to be hydrophobic in nature, due to the absence of any oxygen bonds.
The NiO particle size distribution was centered at 4-5 nm. This good dispersion has been attributed to the interaction between the hydrophilic silica/silicon oxycarbide phases and the nickel-containing precursor salt.18 After sulfidation, the NiS2 particle size distribution was around 20 nm. This enlargement of the nickel particle was in close agreement with the literature, which have reported that the sulfidation process underwent through a very mobile nickel oxysulfide phase.18
Selective oxidation of H2S
Sub-dewpoint processes .The NiS2/SiC catalyst exhibited at 100 ºC a total and stable H2S conversion, together with a selectivity into elemental sulfur of 100 % (no trace of H2S and SO2 at the outlet of the reactor), whereas the solid sulfur deposit on the catalyst surface increased and reached 80 wt. % of the starting weight of the catalyst (Figure 3). This process allows an overall efficiency of sulfur recovery of 100% to be obtained, including the Claus step.20 The high selectivity was attributed to the use of a low reaction temperature in close agreement with the literature, which reported that SO2 formation only occured at higher temperatures. Steijns et al.21 showed that between 100 ºC and 200 ºC, the rate of formation of SO2 by the successive oxidation of sulfur by oxygen was 100 times lower than the rate of oxidation of H2S, due to the very large difference between the corresponding experimental activation energies for oxidation.
The use of a temperature of 100 ºC (below the dewpoint of sulfur) requires a periodical regeneration of the catalyst, due to the continuous deposition of solid sulfur on the catalyst surface during the reaction. Phases of regeneration could be efficiently performed by heating the sulfur loaded catalyst at 300 ºC (over the dewpoint of sulfur) under an inert He flow. It led to the removal of sulfur by vaporizing the sulfur out from the catalyst body and consequently condensing it at the outlet of the catalyst bed. According to the observed results, the regeneration of the sulfur loaded catalysts was complete, as the catalyst recovered its starting weight after the deposited sulfur had been evaporated. No deactivation of the catalyst was observed as a function of cycles of test at 100 ºC and regeneration at 300 ºC (not shown).
The solid sulfur formed by reaction between H2S and oxygen on the NiS2 active particles should very rapidly block the access of the reactants to the active phase, i.e., for low solid sulfur amounts on the catalyst, as usually reported over traditional catalysts.21,22 A peculiar sulfur deposition mode on the catalyst surface has been advanced, in order to explain why the catalyst could maintain high performances, even when the sulfur loading on the catalyst surface reached 80 wt. %. The study of the active phase accessibility was performed by TEM. Figure 3 evidenced that the sulfur loading did not occur directly on the NiS2 active phase, but was located on the b-SiC support, i.e., around the NiS2 particles, the active phase particles also remaining free for the access of the reactants. The formation on stream of a superficial nickel oxysulfide by reaction between the nickel sulfide and oxygen, thus surrounding a NiS2 core, could be proposed to explain the difference in contrast of the NiS2 particle, as it has been proposed by de Jong et al. for MoO3-based catalysts.23 The formation of a superficial nickel oxysulfide phase will be discussed in a further section, the too high reactivity between sulfide and oxygen at 100 ºC avoiding the possible formation of this nickel oxysulfide to be evidenced. It should be mention that a difference in the thickness of the particle could also explain the difference of contrast.
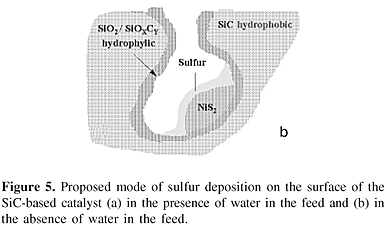
The peculiar mode of sulfur deposition on the catalyst surface proposed, involved the heterogeneous nature of the SiC surface and the water present in the feed. Concerning the heterogeneous nature of the SiC surface reported in the previous section (being partly hydrophilic and hydrophobic), we proposed that the hydrophilic SiOxCy/SiO2 part of the support covered the internal surface of pores, due to the high density of crystal defects in these areas, made of high Miller index planes and easily superficially oxidable. The hydrophobic zones also consisted of low Miller index planes forming the external surface between the pores. More details are reported in Nhut et al.24 The nickel sulfide phase is probably located on the hydrophilic parts in the pore of the catalyst, because of the aqueous impregnation. In the presence of water during the reaction, the formation of a water film on the hydrophilic part of the SiC surface would also occurred by capillarity. This water film could allow the sulfur particles to be continuously removed from the active sites, as a conveyor belt, to the hydrophobic zones outside the pores where the liquid water film stops. No active phase would be located on these zones, and large sulfur particles could be stored without deactivation by active site encapsulation.
When the reaction was performed in dry reaction conditions (not shown), the catalyst rapidly deactivated as a function of time on stream, even for a very low solid sulfur loading on the catalyst surface, similarly to the results reported in the literature over traditional catalysts.18,19 This was attributed to the absence of any liquid film on the surface, which led to the blockage of the active sites by the solid sulfur particles, not mechanically removed.
Figure 5 schematizes the proposed process of sulfur deposition occurring outside the pores of the material when the reaction is performed in the presence of water in the feed (Figure 5a) and inside the porosity in the absence of water in the feed (Figure 5b). On one hand, in the presence of water, the water film acted as a conveyor belt to transport the sulfur particles out of the catalyst pores. On the other hand, in the absence of water, the solid sulfur particles were directly deposited onto the active sites in the pores of the catalyst.
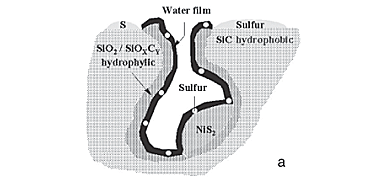
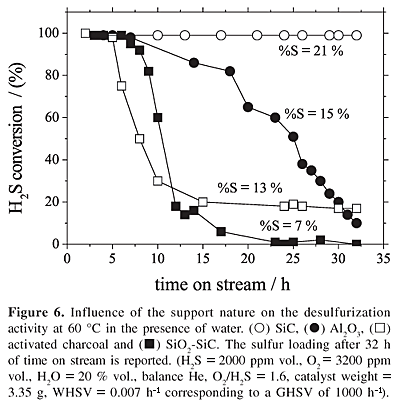
In view of the extremely high activity obtained at 100 ºC, the NiS2/SiC catalyst was tested at a lower temperature, i.e., 60 ºC.25 At this temperature, a large fraction of the steam condensed at the head of the reactor and the reaction was operated in a trickle-bed mode. The formation of the water film on the hydrophilic parts of the SiC surface occurred at 60 ºC directly by water condensation. In order to check the validity of the proposed sulfur deposition mode, the performances of the SiC-based catalyst were compared to the same Ni catalyst supported on pure hydrophilic supports (Al2O3 and SiC oxidized at 1000 ºC for 3 h, leading to a SiO2 coverage, called SiO2-SiC) or activated charcoal (AC), more hydrophobic in nature (Figure 6). The selectivity into elemental sulfur was 100 % for all catalysts. The mixed hydrophilic/hydrophobic SiC-based catalyst remained totally active for large amounts of sulfur stored, whereas the catalysts supported on Al2O3, SiO2-SiC and AC deactivated on stream even with low sulfur loading on their surfaces (see Figure 6). It showed that supports which have only either hydrophilic or hydrophobic zones did not allow to obtain a stable activity as it was obtained using b-SiC with hydrophilic and hydrophobic areas. It confirmed that both hydrophilic and hydrophobic surfaces are required to maintain a high desulfurization activity for a highly sulfur loaded catalyst.
When the reaction was performed at a lower temperature, 40 ºC instead of 60 ºC, the catalyst required a few hours activation period on stream before reaching 100 % conversion (Figure 7). This activation period could be by-passed by a slight activating pretreatment under an oxygen flow at 40 ºC for 1 h and the catalyst was directly active after this oxidative treatment. This activation period, on stream or during the pretreatment, was attributed to the time required by the NiS2 phase to be superficially transformed into a new active phase, i.e. formation of an oxysulfide phase by oxygen and sulfur atom exchange, as reported by de Jong et al.23 The transformation of the supported phase remained superficial, due to the use of a low temperature. The formation of nickel oxide and sulfate has been rejected by comparison with the performances obtained over nickel oxide and sulfate catalysts respectively.18,26 In both cases, the selectivity into sulfur remained total and stable on stream due to the low reaction temperature. The lack of any activation period observed on the test carried out at 60-100 ºC was attributed to the higher reactivity between sulfide and oxygen at 60-100 ºC compared to 40 ºC, which considerably shortened the time needed for the formation of the nickel oxysulfide.
Above-dewpoint process. At above-dewpoint temperatures, Ni must be replaced by Fe because of its high activity, leading to a low selectivity into elemental sulfur by the production of large amounts of SO2.27Figure 8 shows the performances of a silicon carbide supported iron oxide catalyst at 240 ºC. The catalyst exhibited a H2S conversion of 100 % and the SO2 concentration of 1000 ppm at the beginning of the test slowly decreased to about 500-600 ppm after few hours on stream, leading to a sulfur yield of 95 % corresponding to a global sulfur removal efficiency of 99.8 % including the Claus step.
The catalytic results have shown, in correlation with results detailed by Keller et al.,29 that the starting iron phase was subsequently modified under the reactant mixture, probably into an iron oxysulfide or a non-stoichiometric sulfate phase, which was very active for the oxidation of H2S and highly selective towards elemental sulfur. The literature reported that Fe2IIIO3 supported on silica was transformed with time on stream under similar reaction conditions via an Fe2IIIO3.xSO3 (x<2) intermediate into a FeIISO4 phase.30 The authors showed that performances were directly related to the nature of the support. The SiC support being partly covered by silica and silicon oxycarbide phases, on which the active phase is probably dispersed, it is propose that the stabilization of a Fe2IIIO3.xSO3 (x<2) oxysulfide phase similar to that observed by van den Brink,30 could occur during the transformation of Fe2IIIO3 to FeIISO4. It should be noted that the stabilization of a molybdenum oxycarbide on b-SiC has been evidenced during the transformation of MoO3 under hydrocarbon/hydrogen gas mixture, leading to a highly active and selective catalyst for the linear alcane isomerization.31 This highly active and selective molybdenum oxycarbide was not formed when a support with strong interactions with active phase was used, e.g. Al2O3, and in this case reduction to MoO2 was observed. The behavior observed during the first hour on stream (with a selectivity to sulfur of 95 %) was attributed to a short adsorption period of H2S on the surface of the oxide, before the beginning of the oxide to oxysulfide transformation (the same adsorption period has been observed starting from the iron sulfate catalyst supported on SiC, before the sulfate to oxysulfide transformation).18,27
The SiC support material provided the following advantages when compared to classical oxidic or carbon-based supports: (i) a high thermal conductivity is very useful in order to avoid hot spots on the catalyst surface due to the high exothermicity of the considered reaction (DH =- 222 kJ mol-1, corresponding to ca. 70 ºC temperature increase per percent of H2S converted in an industrial reactor). This could lead to a decrease in the selectivity into elemental sulfur by formation of SO2 which is favored at high temperature. A recent review compared the thermal conductivity of SiC to that of some conventional support materials such as alumina or activated carbon;15 (ii) the absence of any microporosity. The microporosity is generally detrimental to the selectivity in the case of selective oxidation applications. Indeed, the presence of micropores in the catalyst led to an "artificial" increase in the residence time of the reacional flow inside the catalytic bed and the catalyst body, thus favoring the successive oxidation of the sulfur formed into SO2. In the present case, at over-dewpoint temperature and in the absence of microporosity, reactants and reaction products were more rapidly evacuated from the catalytic zone, and also hindered the successive oxidation of sulfur onto SO2; (iii) the absence of any acidic and/or basic sites of the b-SiC support, compared to supports such as Al2O3 or SiO2. Indeed, acidic and basic sites have been reported to induce secondary reactions, such as the formation of sulfur radicals leading to SO2 formation, and to promote the Claus equilibrium (equation 1), leading to a decrease in both H2S conversion and sulfur selectivity;6,18,27,29 (iv) the chemical inertness prevents reactions between the reactants, the products and/or the active phase with the support. Most of the traditionally used oxidic supports generally lead to the deactivation of the catalyst with time on stream, due to the chemical reactivity of the supports. The chemical reactivity of the support was reported to lead to a deactivation of the catalyst, because of: (a) the formation of a new compound by reaction between the support and the supported active phase, such a new compound being inactive for the reaction, (b) the sulfation of the support, which can lead to the destruction of the support and/or to encapsulation of the active phase.
SiC prepared according to the Shape Memory Synthesis method was very successfully used as support material for nickel- or iron-based active phases in the direct and selective oxidation of H2S into elemental sulfur in a large range of temperatures. The high performances obtained were notably due to the properties of the SiC support. The efficient replacement of traditionally used supports such as alumina, activated carbon or silica by SiC for several applications, at the laboratory scale or after the technology transfer to the industrial level in close relationship with the industrial partners, leads to hope for a real future use for such a material. For this reason, the newly built SICAT company is now deeply involved in the development of medium surface area silicon carbide-based materials.
Acknowledgement
This work was financially supported by Pechiney (France), Elf Aquitaine (France) and Lurgi Oil (Germany). RV is indebted to CNPq Brazil for the support and financial resources.
Received: December 10, 2003
Published on the web: February 21, 2005
- 1. Estep, J. W.; McBride Jr. G. T.; West, J. R.; Advances in Petroleum Chemistry and Refining, vol.6, Interscience: New York, 1962, ch. 7.
- 2. Wieckowska, J.; Catal. Today 1995, 24, 405.
- 3. "Recent developments to the SCOT process"; Sulphur 1993, 227, 39.
- 4. Savin, S.; Nougayrède, J.; Willing, W.; Bandel. G.; Inter. J. Hydro. Eng. 1998, 54.
- 5. Goar, B. G.; Lagas, J. A.; Borsboom, J.; Heijkoop, G.; Sulphur 1992, 220, 44.
- 6. Pieplu, A.; PhD Thesis, University of Caen, France, 1998.
- 7. Vannice, M. A.; Chao, Y. L.; Friedman, R. M.; Appl. Catal. 1986, 20, 91.
- 8. Lednor, P. W.; Catal. Today 1992, 15, 243.
- 9. Moene, R.; Boon, H. T.; Schooman, J.; Makkee, M.; Moulijn, J. A.; Carbon 1996, 34, 567.
- 10. Boutonet-Kizling, M.; Stenius, P.; Andersson, S.; Frestad, A.; Appl. Catal. B 1992, 1, 149.
- 11. Ledoux, M. J.; Pham-Huu, C.; Chianelli, R. R.; Curr. Opin. Solid State Mater. Sci. 1996, 1, 96.
- 12. Keller, N.; Pham-Huu, C.; Roy, S.; Ledoux, M. J.; Estournès, C.; Guille, J.; J. Mater. Sci. 1999, 34, 3189.
- 13. Yao, H. C.; Stepien, K.; Gandhi, S.; J. Catal. 1980, 61, 547.
- 14. Ledoux, M. J.; Guille, J. L.; Hantzer, S.; Dubots, D.; US pat 4,914,070 1990
- 15. Ledoux, M. J.; Pham-Huu, C.; Cattech 2002, 5, 4, 226.
- 16. Dubots, D.; US pat 5 196 389 1993
-
17SICAT Company. Contact: Dr. Claude Job, 14 avenue Hoche 75008 Paris, France.
- 18. Keller, N.; PhD Thesis, Université Louis Pasteur, Strasbourg, 1999.
- 19. Keller, N.; Pham-Huu, C.; Ledoux, M. J.; Estournès, C.; Ehret, G. ; Appl. Catal. A 1999, 187, 255.
- 20. Philippe, A.; Nougayrède, J. B.; Savin-Poncet, S.; Ledoux, M. J.; Pham-Huu, C.; Crouzet, C.; French pat 94-13752 1994
- 21. Steijns, M.; Derks, F.; Verloop A.; Mars, P.; J. Catal 1976, 42, 87.
- 22. Primavera, A.; Trovarelli, A.; Andreussi, P.; Dolcetti, G.; Appl. Catal. A 1998, 173, 185.
- 23. de Jong, A. M.; Borg, H. J.; van Ijzendoorn, L. J.; Soudant, V. G. M. F.; de Beer, V. H. F.; van Veen, J. A. R.; Niemantsverdriert, J. W.; J. Phys. Chem 1993, 97, 24, 6477.
- 24. Nhut, J. M.; Pesant, L.; Keller, N.; Pham-Huu, C.; Ledoux, M. J.; Top. Catal. 2004, 30, 353
- 25. Ledoux, M. J.; Nougayrède, J. B.; Savin-Poncet, S.; Pham-Huu, C.; Keller, N.; Crouzet, C.; French pat 97-16617, 1997
- 26. Keller, N.; Pham-Huu, C.; Estournès, C.; Ledoux, M. J.; Appl. Catal. A 2002, 234, 193.
- 27. Ledoux, M. J.; Nougayrède, J.-B.; Pham-Huu, C.; Keller, N.; Savin-Poncet, S.; French pat 98-11941 1998
- 28. Keller, N.; Pham-Huu, C.; Ledoux, M. J.; Appl. Catal. A 2001, 217, 205.
- 29. Van den Brink, P. J.; PhD Thesis, University of Utrecht, Netherland, 1992.
- 30. Pham-Huu, C.; Del Gallo, P.; Peschiera, E.; Ledoux, M. J.; Appl. Catal. A 1995, 132, 77.
Publication Dates
-
Publication in this collection
24 May 2005 -
Date of issue
Apr 2005
History
-
Accepted
21 Feb 2005 -
Received
10 Dec 2003