Abstracts
The development and validation of a HPLC method to follow the degradation of phenol by electrochemical or photoelectrochemical treatment are described. This method, which allows determining amounts of phenol, p-benzoquinone and cathecol in electrolyzed or photoelectrolyzed solutions, was used for analysis of reaction intermediates during electrooxidation or photoelectrooxidation of phenol using a Ti-Pt/PbO2 (obtained by thermal-electrochemical or electrodeposition methods) or Ti/TiO2 doped with Ga3+ (obtained by thermal decomposition of precursor salts) electrode.
wastewater treatment; HPLC method; phenol degradation; PbO2 electrodes; Ti/TiO2 photoelectrodes
Descreve-se o desenvolvimento e validação de um método de CLAE para acompanhar a degradação de fenol por tratamento eletroquímico ou fotoeletroquímico. Este método, que permite a determinação das quantidades de fenol, p-benzoquinona e catecol em soluções eletrolisadas ou fotoeletrolisadas, foi usado para análise dos intermediários de reação durante a eletroxidação ou fotoeletroxidação de fenol. Foram usados como eletrodos, Ti-Pt/PbO2 (obtido por método térmico-eletroquímico ou por eletrodeposição) ou Ti/TiO2 dopado com Ga3+ (obtido por decomposição térmica de sais precursores).
ARTICLE
Development of a HPLC method to follow the degradation of phenol by electrochemical or photoelectrochemical treatment
Leonardo S. Andrade; Edison A. Laurindo; Regina V. de Oliveira; Romeu C. Rocha-Filho; Quezia B. Cass * * e-mail: quezia@dq.ufscar.br
Departamento de Química, Universidade Federal de São Carlos, CP 676, 13560-970 São Carlos - SP, Brazil
ABSTRACT
The development and validation of a HPLC method to follow the degradation of phenol by electrochemical or photoelectrochemical treatment are described. This method, which allows determining amounts of phenol, p-benzoquinone and cathecol in electrolyzed or photoelectrolyzed solutions, was used for analysis of reaction intermediates during electrooxidation or photoelectrooxidation of phenol using a Ti-Pt/PbO2 (obtained by thermal-electrochemical or electrodeposition methods) or Ti/TiO2 doped with Ga3+ (obtained by thermal decomposition of precursor salts) electrode.
Keywords: wastewater treatment, HPLC method, phenol degradation, PbO2 electrodes, Ti/TiO2 photoelectrodes
RESUMO
Descreve-se o desenvolvimento e validação de um método de CLAE para acompanhar a degradação de fenol por tratamento eletroquímico ou fotoeletroquímico. Este método, que permite a determinação das quantidades de fenol, p-benzoquinona e catecol em soluções eletrolisadas ou fotoeletrolisadas, foi usado para análise dos intermediários de reação durante a eletroxidação ou fotoeletroxidação de fenol. Foram usados como eletrodos, Ti-Pt/PbO2 (obtido por método térmico-eletroquímico ou por eletrodeposição) ou Ti/TiO2 dopado com Ga3+ (obtido por decomposição térmica de sais precursores).
Introduction
In the recent past, problems involving water contamination have called the attention to the necessity of removing toxic organic compounds from industrial aqueous effluents. Phenolic compounds are found in high concentrations in the residual waters of petrochemical, paint, paper and textile industries. These compounds are toxic to the environment; for example, phenol in concentrations higher than 2 mg L-1 is considered toxic to fish and, taking into account four days of exposure, concentrations between 10 mg L-1 and 100 mg L-1 are lethal to most aquatic life.1 Thus, phenolic compounds must be removed before disposition of the residual waters can be carried out.
For this removal, use of biological methods may be a satisfactory solution. However, there are many compounds for which these methods can be inefficient or require long-term treatment in order to open the aromatic ring; in some instances, this can take years to be accomplished.2 In such cases, electrochemical and photoelectrochemical methods have been considered as a good alternative, since most aromatic compounds can thus be transformed into biodegradable substances or, better yet, depending on the electrode material used, complete oxidation (often referred as combustion) can be accomplished by electrochemical3 or photoelectrochemical4 methods.
As it is shown in Scheme 1, the first step in the anodic oxidation of phenol involves the formation of the phenoxy radical with the para and orto positions activated.5 After, the aromatic ring is hydroxylated to form hydroquinone and/or cathecol followed by p-benzoquinone and/or o-benzoquinone formation. Finally, the ring is opened to form biodegradable aliphatic acids that are then mineralized.
Most of the papers that report on the oxidation of phenolic compounds do not take into account (or do not mention) the necessity of analytical criteria associated to the HPLC measurements of the compounds formed in the oxidation process; in other words, no method validation is presented (or mentioned). Thus, the objective of this work was to develop and validate a HPLC method for the analysis of reaction intermediates during the electrooxidation of phenol using Ti-Pt/PbO2 electrodes (obtained by the thermal-electrochemical6 method or by electrodeposition7), or the photoelectrooxidation of phenol using a Ti/TiO2 doped with Ga3+ (10 cmol mol-1) electrode, obtained by thermal decomposition of precursor salts. Phenol and the intermediate compounds hydroquinone, p-benzoquinone and cathecol were selected for the development of this method. The validated method was used for the kinetic studies of the oxidation processes.
Experimental
Chemicals
Phenol (Merck®) and p-benzoquinone, hydroquinone, cathecol (Acros®) were used as standards. Methanol and tetra-hydrofurane THF (HPLC grade) were obtained from Mallinckrodt® and J. T. Baker®, respectively. Water was purified using a Milli-Q system (Millipore). Buffer solutions used as part of the mobile phase were prepared using sodium monohydrogenphosphate (Merck®) and phosphoric acid (Merck®).
Equipment
A Shimatzu HPLC system, consisting of a SIL 10AF auto-injector, a SPD 10A UV detector operated at 254 nm, a SCL 10A control system and a LC 10AT pump, was used. The data acquisition was carried out using the Class LC10 software (Shimatzu).
Chromatographic conditions
The mobile phase used was a mixture of 0.01 mol L-1 potassium monohydrogenphosphate solution (pH adjusted to 3 with H3PO4), methanol and THF in the volumetric proportion of 90:5:5. The flow rate used was 1.0 mL min-1 and detection was monitored at l = 254 nm.
The analytical column (4.6 mm diameter and 150 mm length) used was packed with C18 nucleosil silica (5 mm, 100 Å; Macherey-Nagel, Germany) by the ascending slurry method, using methanol for the preparation of the slurry (50 mL) and also for the packing. This was carried out at a pressure of 7500 psi; after, the column was conditioned for about 4 h with methanol at a flow rate of 1.0 mL min-1.8
Stock solutions
Stock solutions of the mixture of compounds used (phenol, p-benzoquinone, hydroquinone and cathecol) were prepared in a concentration of 2 g L-1 of each compound using the mobile phase as solvent. These solutions were used to prepare the calibration standards with the following nominal concentrations: 2.4 mg L-1, 4.8 mg L-1, 10 mg L-1, 20 mg L-1 and 30 mg L-1.
Method validation
The solutions were prepared in triplicate and transferred to the autosampler vials and samples of 100 mL were injected into the column. Calibration curves were constructed by plotting the peak area against the nominal concentration of each compound.
Five samples of each compound were prepared at the quality control concentrations of 2.8 mg L-1, 15 mg L-1 and 25 mg L-1, in three different days, in order to determine intra- and inter-day precision and accuracy.
The acceptance criteria for the limit of quantification was that the accuracy for three analyzed samples presented less than 15% variability and the limit of detection was measured taking into account a signal-to-noise ratio of three.
Kinetic study
The reaction solutions were prepared by dissolving 50 mg of phenol in 100 mL of a 1.0 mol L-1 H2SO4 solution. Analyses of the samples were carried out at time zero and then at each hour for a period of seven hours. For the analysis, an aliquot (20 µL) of the reaction media was diluted with 380 µL of the mobile phase and 250 µL was transferred to the auto-sampler vials and samples of 100 µL were injected in the analytical column.
Results and Discussion
The main intermediates during the electrochemical/photoelectrochemical degradation of phenol are hydroquinone, cathecol and 1,4-benzoquinone;5 thus these compounds were selected for the method development.
Reversed-phase elution mode, using a Nucleosil® C18 column, was investigated under isocratic conditions to achieve the necessary retention of the four compounds. Phosphate buffer (0.01 mol L-1) with pH adjusted to 3 with phosphoric acid, in methanol (80:20, v/v) was initially used. The low pH was necessary for increasing the retention time of the ionized compounds and improving peak shape. To control the selectivity, a wide range of mobile phases was investigated and the use of acetonitrile, THF and methanol (or a mixture of these solvents) as modifier was evaluated. Good selectivity and retention factors were obtained when a 0.01 mol L-1 phosphate buffer (pH adjusted to 3 with phosphoric acid) with methanol and THF (90:5:5, v/v) was used as the mobile phase at a flow rate of 1 mL min-1.
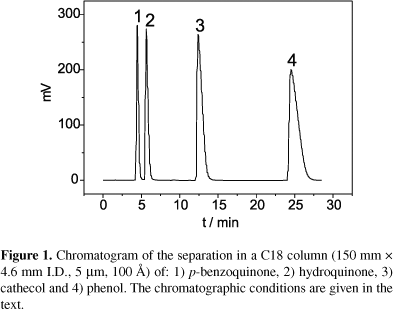
The chromatogram in Figure 1 illustrates the separation achieved under these conditions and shows also the elution order, which was identified by co-injection of the individual compounds.
The response of the UV detector at 254 nm was linear for each compound, from 2.4 mg L-1 to 30 mg L-1. Least-squares regression analyses of the data for all four compounds resulted in excellent straight-line fits over the concentration examined, with R2 superior to 0.999. Figure 2 shows the calibration curves obtained.
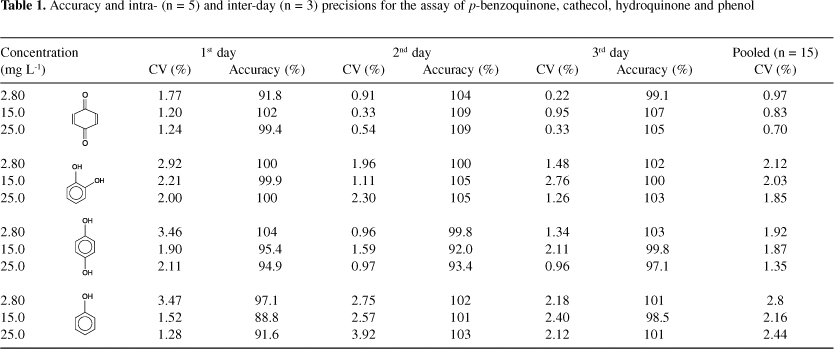
The intra and inter-day precision and accuracy of the method were determined by analyzing five replicates of three quality controls on three non-consecutive days. The precision was expressed as coefficients of variation (CV%) and the accuracy was evaluated by back-calculation and expressed as the percent deviation between amount found and amount added at the three concentrations examined. The obtained results, which led to satisfactory intra- and inter-assay precision and accuracy, are shown in Table 1.
The limit of quantification found for all four compounds was of 1.2 mg L-1 while the calculated limits of detection were 0.075 mg L-1 for phenol, cathecol and hydroquinone, and 0.0003 mg L-1 for benzoquinone.
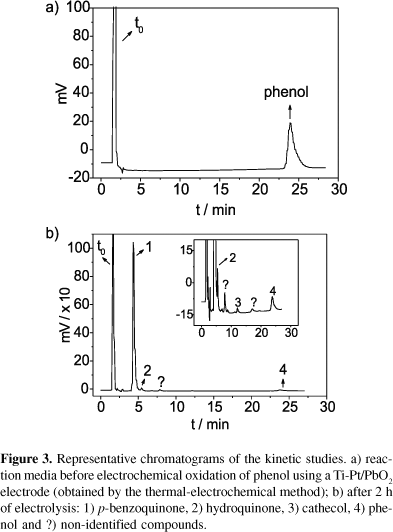
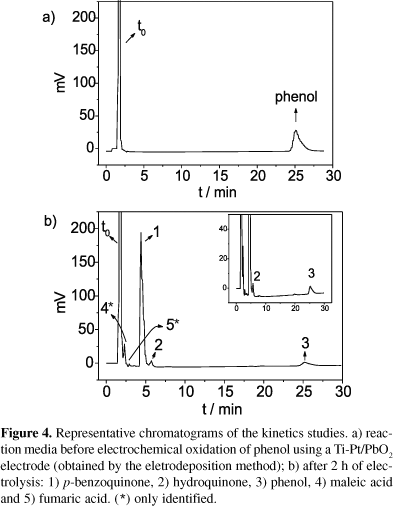
The developed method showed good linearity, sensitivity, selectivity, intra and inter-day precision and accuracy and, thus, was used to follow the electrochemical degradation of phenol under different experimental conditions. The intermediate compounds formed were quantified and the results obtained in these experiments are given in Tables 2 and 3. The chromatograms shown in Figure 3 and 4 are representative of samples analyzed to evaluate the method applicability using the two Ti-Pt/PbO2 electrodes obtained by different methods. Tables 2 and 3 show the corresponding results for the degradation of phenol. Under photoelectrochemical conditions (using the Ti/TiO2 doped with Ga3+ electrode), out of the four compounds only benzoquinone was formed, but below quantification level; these results are shown in Table 4.
The different performances of the two PbO2 anodes, showed in the Tables 2 and 3, can be explained taking into account their electroactivity for the oxygen evolution reaction (OER). The electrode obtained by the thermal-electrochemical method (TM) has a lower OER overpotential than that obtained by the electrodeposition method. X-ray diffractograms allowed the characterization of the thermal-electrochemical oxide as a-PbO2 and of the electrodeposited oxide as b-PbO2. Electrodes with low OER overpotential favor selective oxidations (conversion) of organics compounds while those with a high overpotential favor the electrochemical combustion.9 Therefore, more reaction intermediate compounds are expected when using the PbO2 electrode obtained by TM and lower concentrations are expected when using the electrodeposited PbO2 electrode. It should be pointed out that ~67% (Figure 3) and 100% (Figure 4) of the reaction intermediate compounds were identified when using the PbO2 (TM) and PbO2 (electrodeposition) electrodes, respectively. Furthermore, in both cases CO2 was quantified using the Total Organic Carbon (TOC) technique.
Conclusions
The resolution obtained under isocratic elution allowed the accurate determination of the intermediate compounds in the phenol electrochemical degradation process. The validated method is simple and rapid, requiring a total analysis time of 26 min per sample and was efficiently used in the kinetic study of these reactions.
Acknowledgments
The financial support (mainly scholarships) of CNPq and FAPESP is gratefully acknowledged.
Received: May 2, 2005
Published on the web: February 17, 2006
FAPESP helped in meeting the publication costs of this article.
- 1. Taken, R. L; Lewis, J., eds.; Registry of Toxic Effects of Chemical Substances; NIOSH (publication nş 83-107): Washington, 1993, pp. 83-107.
- 2. Rajeshwar, K.; Ibanez, J. G.; Swain, G. M.; J. Appl. Electrochem. 1994, 24, 1077.
- 3. Comninellis, C.; Electrochim. Acta 1994, 39, 1857.
- 4. Rajeshwar, K.; Ibanez, J. G.; Environmental Electrochemistry: Fundamentals and Applications in Pollution Abatement, Academic Press: S. Diego, 1997.
- 5. Gattrell, M.; Kirk, D. W.; J. Electrochem. Soc. 1993, 140, 1534.
- 6. Laurindo, E. A.; Bocchi, N. ; Rocha-Filho, R. C.; J. Braz. Chem. Soc. 2000, 11, 429.
- 7. Bemelmans, C.; O'Keef, T.; Colie, E.; Bull. Electrochem. 1996, 12, 591.
- 8. Cass, Q. B.; Gomes, R. F.; Calafatti, S. A.; Pedrazolli Jr., J.; J. Chromatogr., A 2003, 987, 235.
- 9. Comninellis, C.; Electrochim. Acta 1994, 39, 1857.
Publication Dates
-
Publication in this collection
17 Apr 2006 -
Date of issue
Apr 2006
History
-
Received
02 May 2005