Abstracts
An efficient, regioselective synthesis of some 2-aroylimino-3-aryl-thiazolidin-4-ones (2a-j) involving base-catalyzed cyclization of 1-aroyl-3-aryl thioureas with chloroacetyl chloride in dioxane is reported. The structures were confirmed by spectroscopic data, elemental analyses and in one case (2j) by single crystal X-ray diffraction data. Compounds (2a-j) were assayed in vitro for their antimicrobial activity against Gram positive and Gram negative bacteria and were found to exhibit promising activity towards the tested microorganisms, comparable to and in some cases better than those of the standard drugs.
iminothiazolidin-4-one; 1-aroyl-3-aryl-thioureas; crystal structure; antibacterial
Este trabalho relata uma síntese eficiente e regio-seletiva de algumas 2-aroylimino-3-aryl-thiazolidin-4-ones (2a-j) envolvendo a ciclização de 1-aroyl-3-aryl tio-uréias em meio básico com cloreto de cloroacetila em dioxana. As estruturas foram confirmadas por dados espectroscópicos, análises elementares e, em um caso (2j), por dados de difração de raios X tipo cristal único. Os compostos foram testados in vitro quanto à sua atividade antimicrobiana em relação a bactérias Gram positivas e Gram negativas. Os resultados revelaram uma atividade promissora desses compostos em relação aos microorganismos testados, comparando-se e, em alguns casos, até superando a atividade das drogas existentes.
ARTICLE
Synthesis and antibacterial activity of some novel 2-Aroylimino-3-aryl-thiazolidin-4-ones
Aamer SaeedI, * * e-mail: aamersaeed@yahoo.com ; Naeem AbbasI; Ulrich FlörkeII
IDepartment of Chemistry, Quaid-I-Azam University, Islamabad 45320, Pakistan
IIDepartment Chemie, Fakultat fur Naturwissenschaften, Universitat Paderborn, Warburgerstrasse 100, D-33098 Paderborn, Germany
ABSTRACT
An efficient, regioselective synthesis of some 2-aroylimino-3-aryl-thiazolidin-4-ones (2a-j) involving base-catalyzed cyclization of 1-aroyl-3-aryl thioureas with chloroacetyl chloride in dioxane is reported. The structures were confirmed by spectroscopic data, elemental analyses and in one case (2j) by single crystal X-ray diffraction data. Compounds (2a-j) were assayed in vitro for their antimicrobial activity against Gram positive and Gram negative bacteria and were found to exhibit promising activity towards the tested microorganisms, comparable to and in some cases better than those of the standard drugs.
Keywords: iminothiazolidin-4-one, 1-aroyl-3-aryl-thioureas, crystal structure, antibacterial
RESUMO
Este trabalho relata uma síntese eficiente e regio-seletiva de algumas 2-aroylimino-3-aryl-thiazolidin-4-ones (2a-j) envolvendo a ciclização de 1-aroyl-3-aryl tio-uréias em meio básico com cloreto de cloroacetila em dioxana. As estruturas foram confirmadas por dados espectroscópicos, análises elementares e, em um caso (2j), por dados de difração de raios X tipo cristal único. Os compostos foram testados in vitro quanto à sua atividade antimicrobiana em relação a bactérias Gram positivas e Gram negativas. Os resultados revelaram uma atividade promissora desses compostos em relação aos microorganismos testados, comparando-se e, em alguns casos, até superando a atividade das drogas existentes.
Introduction
The extensive use of antibiotics has led to the appearance of multi-drug resistant microbial pathogens.1 This highlights the incessant need for the development of new classes of antimicrobial agents and alteration of known drugs in such way that would allow them to retain their physiological action, but reducing their resistance to the pathogen. The design of novel chemotherapeutic agents is particularly beneficial due to their dissimilar mode of action which can avoid cross resistance to known drugs.
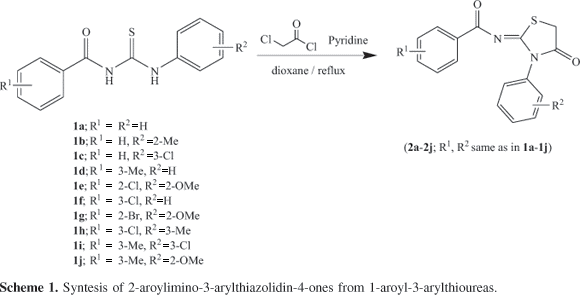
There has been considerable interest in the chemistry of thiazolidin-4-one ring systems, which is a core structure in various synthetic pharmaceuticals displaying a broad spectrum of biological activities.2 Thiazolidin-4-one ring also occurs in nature; thus actithiazic acid [() 2-(5-carboxypentyl) thiazolidin-4-one)] isolated from Streptomyces strains exhibits highly specific in vitro activity against Mycobacterium tuberculosis. 3 Thiazolidin-4-one derivatives are known to exhibit diverse bioactivities such as anticon-vulsant,4 antidiarrheal,5 anti-platelet activating factor,6 antihistaminic,7 antimicrobial,8 antidiabetic,9 cycloxygenase inhibitory,10 Ca2+ channel blocker,11 PAF antagonist,12 cardioprotective,13 anti ischemic,14 anti cancer,15 anti HIV,16 non-peptide thrombin receptor antagonist17 and tumor necrosis factor-a antagonist activities.18 However, the 2-imino derivatives of thiazolidinones are explored to a lesser extent, in spite of the fact that the presence of imino function provides an additional point of structural diversity in a thiazolidinone-based pharmacophore (Figure 1). It may be due to the lack of efficient synthetic access to iminothiazolidinones, therefore, design of a general, simple and efficient method for rapid synthesis of thiazolidine-4-ones would be greatly valuable and could warrant further investigations in drug discovery. Recently, different protocols have been developed allowing the synthesis of imino thiazolidin-4-one skeletons.19-21 In this connection, herein we report the synthesis of some new 2-iminothiazolidin-4-ones and their biological screening. Ten 2-aroylimino-3-aryl-thiazolidin-4-ones (2a-j) were synthesized and tested for their in vitro antimicrobial properties against Gram positive and Gram negative bacteria. All the new compounds (2cj) were characterized by physical data, elemental analyses and spectroscopic data (1H and 13C NMR, MS and IR) and in case of 2j by single crystal X-ray diffraction data.
Results and Discussion
Chemistry
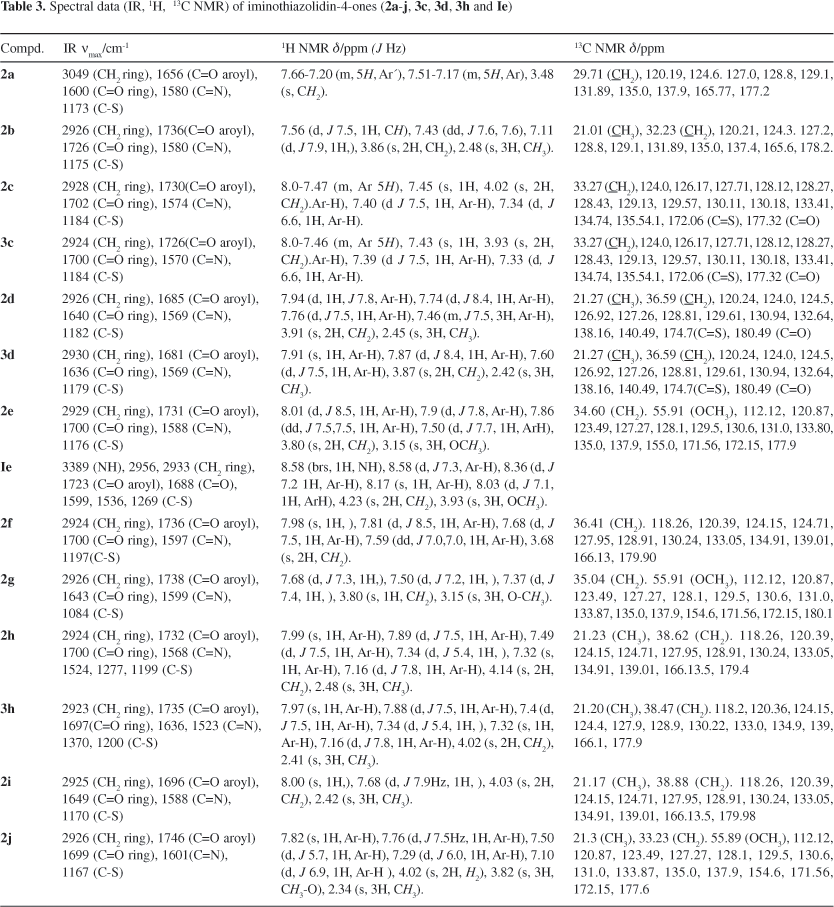
1-Aroyl-3-arylthiourea derivatives (1aj) were synthesized using procedure reported earlier,22 starting from reaction of suitable aroyl chlorides with potassium thiocyanate in acetone followed by treatment with appropriate anilines. Typically, aroyl thioureas are characterized by IR absorptions at 3350-3320, 3250-3200 for the free and associated NH, at 1650-1670 for carbonyl and at 1230-1250 cm-1 for thiocarbonyl groups respectively. The characteristic broad singlets at ca. d 9 and 12 for HN(1) and HN(3) and peaks at ca. d 170 and 179 for carbonyl and thiocarbonyl were observed in the 1H and 13CNMR spectra respectively.
The next step involving the base-catalyzed cyclization of 1-aroyl-3-arylthioureas with chloroacetyl chloride may lead to different condensation products by S or N intramolecular cyclization by a slight variation of conditions; therefore, it is important to first unearth the set of optimized conditions. The results of reaction optimization under different conditions varying the solvent, the reagent and the base are summarized in Table 1 using 1j as a model compound. It can be seen that the use of a strong base like Et3N or polar aprotic solvent like DMF noticeably reduced the reaction time (entries 1, 2 vs. 3) but leads to a mixture of regioisomers 2 and 3 obtained by initial S-attack followed by N1 or N3 cyclization besides, the N-attack cyclization products (thiohydantoins).23 By applying the optimized conditions, involving use of chloroacetyl chloride, dry dioxane as solvent in the presence of pyridine as base (entry 4) the 2-imino-4-thiazolidinones with various substituents (2a-j) were synthesized (Scheme 1) from the corresponding 1-aroyl-3-aryl thioureas (1a-j) in good yields (Table 2). Under these conditions, in majority of the cases a ratio of 9:1 was obtained for regioisomers 2 and 3 as shown by the relative intensities of their signals in the 1H NMR spectra and/or separation of 3 by thick layer chromatography (Table 2). The yields, the physicochemical properties and the spectroscopic data of 2a-j are given, respectively, in Tables 2 and 3. The compounds were characterized by the absence of both NH absorptions of the thiourea moiety and the appearance of the characteristic C=O and C=N absorptions of the thiazolidin-4-one ring in the regions 1680-1700 and 1560-1590 cm-1, respectively in addition to the ArC=O, C=C and CS absorptions in the IR spectra. The 1H NMR spectra of compounds 2a-j displayed the characteristic singlet for CH24 methylene group deshielded by the adjacent C=O and sulfur atom, at d 3.8-4.3 and 13C NMR showed the analogous peaks at d 29-33, besides the signals for aromatic protons. In the mass spectrum, in addition to the molecular ion peaks, the base peaks originated from the aroyl cation fragment was observed. The regioisomer 3 was also isolated and characterized spectroscopically in some of the cases. The regioisomers 2 and 3 have closely related Rf values and parallel set of the characteristic signals in the IR and NMR spectra. In general, regioisomer 2 has slightly lower Rf but slightly higher d values compared to those of 3 (Table 3). Thus, in a typical case the characteristic methylene singlets were found at d 4.14 and 4.02 for 2h and 3h receptively in the NMR and the absorptions for aroyl and ring carbonyls were found at 1732, 1707 and 1735, 1697 cm-1 receptively in the IR spectra.
The mechanism of cyclization involves the attack of sulfur in the resonance stabilized anion generated by loss of proton from N-1 by base, to the a-carbon of the chloroacetyl chloride followed by the attack of anion from N-3 to the carbonyl of chloroacetyl intermediate species (I) and ensuing cyclization to yield (2) as the predominant regioisomer (Scheme 2). The formation of acyclic intermediate of type I is supported by the NMR and IR spectra of a spot of decreasing intensity with time in the reaction mixture having Rf value in between those of the thiourea and the cyclized product(s). Thus, in case of Ih the broad singlet at d 12.0 for NH-1 had disappeared while that at d 9.0 was intact and singlet for a methylene group flanked by carbonyl and sulpher atom was also observed at d 4.23, in the 1H NMR and similar changes were noticed in IR spectrum. The formation of less stable regioisomer 2-arylimino-3-aroyl-thiazolidin-4-ones 3 can be visualized by a similar pathway initiating from S-attack followed by cyclization from the direction of less stable anion (N-3) (Scheme 3). The predominance of isomer 2 over 3 can be explained by the extended conjugative stabilization of aroyl carbonyl with the imine and ring nitrogen. The regiocontrol in the cyclization of thioureas bearing an aroyl and an aryl substituent is typically influenced by thermodynamic stability of regioisomer 2 over 3. This has also been supported by the bond length of 1.3775 for N2-C9 compared to a bond length of 1.3827 for N2-C11 showing a partial double bond nature of the former.
X-ray data collection, structure solution and refinement.
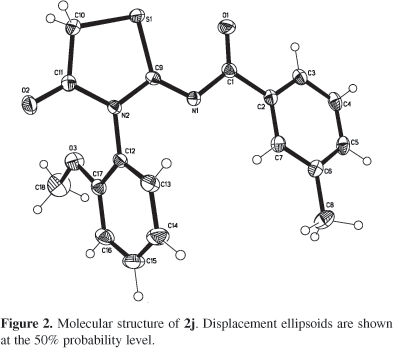
The regiochemistry of 2 was established unequivocally by single crystal X-ray analysis24 (Figure 2). C18H16N2O3 S, Mr = 340.4, monoclinic, space group P 21/c, a = 7.5665(4), b = 22.1951(13), c = 10.0762(6) Å, b = 90.198(1)°, V = 1692.2(2) ų, Z = 4, Dx = 1.336 g cm-³, F(000) = 712, T = 120(2) K. Bruker-AXS SMART APEX CCD diffractometer,25 graphite monochromator, l (MoKa) = 0.71073 Å, µ = 0.209 mm-1, colourless prismatic crystal, size 0.43 ´ 0.40 ´ 0.36 mm3, 13091 intensities collected 2.22 < q < 28.08°, 10 < h < 8, 29 < k < 29, 13 < l < 13. Structure solved by direct methods,25 full-matrix least-squares refinement25 based on F2 and 217 parameters, all but H atoms refined anisotropically. H atom positions determined from DF maps and refined at idealized positions riding on the carbon atoms with isotropic displacement parameters Uiso(H) = 1.2U(Ceq) or 1.5U(CH3). All CH3 groups were allowed to rotate but not to tip. Refinement converged at R1(F) = 0.041, wR2(F2, all data) = 0.110, S = 1.033, max(d/s) < 0.001, min/max height in final DF map 0.20/0.42 e/Å3.
The molecular structure of 2j was established by single crystal X-ray analysis (Figure 2). As one might predict based on sterical considerations, the methoxybenzyl group is oriented away from the NCSC2 plane with a dihedral angle of 71.67(4)°. The methoxy group lies in the plane of the benzene ring, the respective torsion angle C18-O3-C17-C12 measures 173.7(1)°. The planes of both aromatic rings are nearly perpendicular making a dihedral angle of 84.83(5)°.Torsion angles C1-N1-C9-S1 2.2(2)° and C9-N1-C1-O1 1.9(2)° indicate the non-twisted conformation. The crystal packing (Figure 3) shows intermolecular hydrogen bridges C5-H..O2 (x-1,+y,+z-1) with CO 3.216(2) Å, C-H..O 138.0(1)° and C14-HO2 (x-1,+y,+z) with CO 3.486(2) Å, C-HO 163.7(1)° that link molecules to endless chains along [100].
Biology
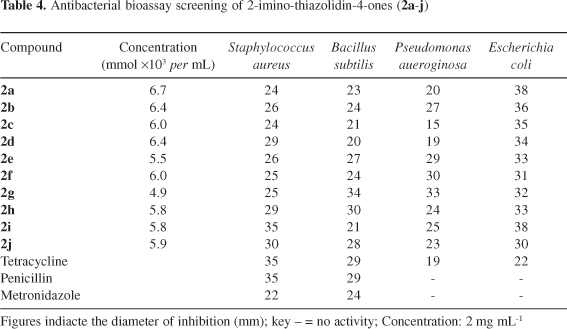
In vitro evaluation of antibacterial activity was carried out by disk diffusion method (Kirby-Bauer method) against different bacterial strains.26 The tests were repeated thrice and the results are reported as averages of at least three determinations. Antibacterial activity of the compounds tested is shown in Table 4. The figures represent the zone of inhibition in millimeters. All of the compounds 2a-2i exhibited promising inhibitory activity against the four bacterial strains compared to standard drugs at the tested concentrations. The presence of halogen groups results in enhancement of inhibitory activity. It may also be interesting to compare the activity of isomeric compounds 2h and 2i having 3-chloro and 3-methyl substituents interchanged on the aroyl and aryl rings.
Conclusion
An efficient cyclization protocol for 1-aroyl-3-aryl thioureas with chloroacetyl chloride to novel 2-aroylimimnothiazolidin-4-ones has been developed. Regiochemistry is controlled by relative stability of the two regioisomers 2 and 3 with 2 as the predominant isomer. The simplicity of the experimental procedure and high yields render this approach particularly attractive. From antimicrobial activity data, it is revealed that, the compounds 2c-i-may serve as promising antimicrobial agents for therapeutic use. However, further biological evaluation, including the determination of MIC values is essentially required for the series of promising compounds.
Experimental
Melting points were recorded using a digital Gallenkamp (SANYO) model MPD.BM 3.5 apparatus and are uncorrected. 1H NMR and 13CNMR spectra were determined in CDCl3 at 300 and 100 MHz respectively using a Bruker AM-300 spectrometer. FTIR spectra were recorded on an FTS 3000 MX spectrophotometer. Mass Spectra (EI, 70eV) on a MAT 312 instrument, and elemental analyses were conducted using a LECO-183 CHNS analyzer. Bioactivities were carried out at the department of microbiology, Quaid-I-Azam University Islamabad. Thin layer chromatography (TLC) was conducted on 0.25 mm silica gel plates (60F254, Merck). Visualization was made with ultraviolet light. Reagents were obtained commercially and used as received.
General procedures for the synthesis of 2-aroylimino-3-arylthiazolidin-4-ones (2aj)
To a stirred solution of 1-aroyl-3-arylthiourea (0.01 mol) in pyridine and 20 mL dry dioxane was added, dropwise, chloroacetyl chloride (1 mmol) under nitrogen and the reaction mixture was refluxed for 0.5-2h. After the reaction was completed the contents were cooled and poured into ice-cold water. The solid mass was separated, filtered, washed with water and then purified by recrystallization from suitable solvents.
The physicochemical and spectral data are given in tables 2 and 3 respectively. All compounds gave satisfactory elemental analyses.
Acknowledgments
We are very grateful to financial grant by URF, Quaid-I-Azam University Islamabad under Project No: DFNS/ 2007-382.
References
1. Andersson, I.; T. van Scheltinga, A. C.; Valegard, K.; Cell; Mol. Life Sci. 2001, 58, 1897; Frere, J.-M.; Mol. Microbiol. 1995, 16, 385.
2. Vigorita, M. G.; Ottana, R.; Monforte, F.; Maccari, R.; Trovato, A.; Monforte, M. T.; Taviano, M. F.; Bioorg. Med. Chem. Lett. 2001, 11, 2791; Chande, M.S.; Suryanarayan, V.; J. Chem. Res. 2005, 6, 345; Kavitha, C.V.; Basappa, S. Swamy, N.; Mantelingu, K.; Doreswamy, S.; Sridhar, M.A.; Prasad S.; Rangappa, K. S.; Bioorg. Med. Chem., 2006, 14, 2290; Shiradkar, M.; Shivaprasad, H.N.; Asian J. Chem. 2006, 18, 331.
3. Sobin, B. A.; J. Am. Chem. Soc. 1952, 74, 2947; Grundy, W. E.; Whitman, A. I.; Rdzok, E. G; Rdzok, E. J.; Haris, M.E. Sylvester, W.E.; Antibiot. Chemother. 1952, 2, 399.
4. Ergene, N.; Capan, G.; Il Farmaco 1994, 49, 449.
5. Diurno, M. V.; Mazzoni, O.; Izzo, A. A.; Bolognese, A.; Il Farmaco 1997, 52, 237.
6. Koike, H.; Imanashi, N.; Natsume Y.; Morooka, S.; Eur. J. Pharm. Mol. Pharm. 1994, 269; Tanabe, Y.; Yamamoto, H.; Murakami, M.; Yanagi, K.; Kubota, Y.; Okumura, H.; Sanemitsu, Y.; Suzukamo, G.; J. Chem. Soc. Perkin Trans. I 1995, 7, 935; Tanabe, Y.; Komuro, Y.; Imanishi, N.; Morooka, S.; Enomoto, M.; Kojima, A.; Sanemitsu, Y.; Mizutani, M.; Tetrahedron. Lett. 1991, 32 379.
7. Diurno, M. V.; Mazzoni, O.; Correale, G.; Monterry, I. G.; Il Farmaco 1999, 54, 579; Previtera, T.; Vigorita, M. G.; Bisila, M.; Orsini, F.; Benetolla, F.; Bombieri, G.; Eur. J. Med. Chem. 1994, 29, 317.
8. Desai, S. B.; Desai, P. B.; Desai, K. R.; Asian J. Chem. 1999, 2, 363; Sharma, R. C.; Kumar, D.; J. Indian Chem. Soc. 2000, 77, 492; Piscapo, E.; Diurno, M. V.; Gagliardi, R.; Mazzoni, O.; Boll. Soc. Ital. Biol. Sper. 1989, 65, 853.
9. Ueno, H.; Oe, T.; Snehiro, I.; Nakamura, S. US Patent 5594116, 1997, Chem. Abstr. 126 (1977) 157507p.
10. Ottaná, R.; Mazzon, E.; Dugo, L.; Monforte, F.; Maccari, R.; Sautebin, L.; De Luca, G.; Vigorita, M. G.; Alcaro, S.; Ortuso, F.; Eur. J. Pharmacol. 2002, 448, 71.
11. Hara, A.; Suzuki, T.; Hashizume, H.; Shishido, N.; Nakamura, M.; Ushikubi, F.; Abiko, Y.; Eur. J. Pharmacol. 1999, 385, 81; Kato, T.; Ozaki, T.; Tamura, K.; J. Med. Chem. 1999, 42, 3134.
12. Tanabe, Y.; Suzukamo, G.; Komuro, Y.; Imanishi, N.; Morooka, S.; Enomoto, M.; Kojima, A.; Sanemitsu, Y.; Mizutani, M.; Tetrahedron Lett. 1991, 32, 379.
13. Kato, T.; Ozaki, T.; Ohi, N. Tetrahedron: Asymmetry 1999, 10, 3963.
14. Adachi, Y.; Suzuki, Y.; Homma, N.; Fukazawa, M.; Tamura, K.; Nishie, I.; Kuromaru, O.; Eur. J. Pharmacol. 1999, 367, 267.
15. Ebeid, M, Y.; Fathallah, O, A.; El-Zaher, M. I.; Kamel, M, M.; Abdon, W, A.; Anwar, M. M.; Bull. Fac. Pharm. 1996, 34 125.
16. Rawal, R.K.; Prabhakar, Y.S.; Katti S.B.; De Clercq E.; Bioorg. Med. Chem. 2005, 13, 6771.
17. Kato, Y.; Kita, Y.; Nishio, M.; Hirasawa, Y.; Ito, K.; Yamanaka, T.; Motoyama, Y.; Seki, J.; Eur. J. Pharmacol. 1999, 384, 197.
18. Voss, M. E.; Carter, P.H.; Tebben, A. J.; Scherle, P. A.; Brown, G. D.; Thompson, L, A.; Xu, M.; Lo, Y, C.; Yang, Liu, R. R.-Q; Bioorg. Med. Chem. Lett. 2003, 13, 533.
19. Vicini, P.; Geronikaki, A.; Anastasia, K.; Incerti, M.; Zani, F.; Bioorg. Med. Chem. 2006, 14, 3859.
20. Blanchet, J.; Zhu J.; Tetrahedron Lett. 2004, 45, 4449.
21. Laurent, D. R. St.; Gao, Q.; Wu, D.; Serrano-Wu, M. H.; Tetrahedron Lett. 2004, 45, 1907.
22. Saeed, A.; Parvez, M.; Cent. Eur. J. Chem. 2005, 3, 780; Saeed A.; Flörke, U.; Acta Cryst. 2006, E62, o2403; Saeed, A.; Flörke, U.; Acta Cryst. 2006, E62, o2530.
23. Durust Y.; Nohut F.; Synth. Commun. 1999, 29, 1997.
24. Full crystallographic data (excluding structure factors have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-616464. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44)1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).
25. Bruker (2002). SMART (Ver. 5.62), SAINT (Ver. 6.02), SHELXTL (Ver. 6.10) and SADABS (Version 2.03). Bruker AXS Inc., Madison, Wisconsin, USA.
26. Cappuccino, J.G.; Sherman, N. In Microbiology: a Laboratory Manual, pp. 247-251, The Benjamin/Cummings publishing Co.: California (USA) (1996).
Received: August 23, 2006
Web Release Date: April 27, 2007
- 1. Andersson, I.; T. van Scheltinga, A. C.; Valegard, K.; Cell; Mol. Life Sci. 2001, 58, 1897;
- Frere, J.-M.; Mol. Microbiol. 1995, 16, 385.
- 2. Vigorita, M. G.; Ottana, R.; Monforte, F.; Maccari, R.; Trovato, A.; Monforte, M. T.; Taviano, M. F.; Bioorg. Med. Chem. Lett. 2001, 11, 2791;
- Chande, M.S.; Suryanarayan, V.; J. Chem. Res. 2005, 6, 345;
- Kavitha, C.V.; Basappa, S. Swamy, N.; Mantelingu, K.; Doreswamy, S.; Sridhar, M.A.; Prasad S.; Rangappa, K. S.; Bioorg. Med. Chem., 2006, 14, 2290;
- Shiradkar, M.; Shivaprasad, H.N.; Asian J. Chem. 2006, 18, 331.
- 3. Sobin, B. A.; J. Am. Chem. Soc 1952, 74, 2947;
- Grundy, W. E.; Whitman, A. I.; Rdzok, E. G; Rdzok, E. J.; Haris, M.E. Sylvester, W.E.; Antibiot. Chemother. 1952, 2, 399.
- 4. Ergene, N.; Capan, G.; Il Farmaco 1994, 49, 449.
- 5. Diurno, M. V.; Mazzoni, O.; Izzo, A. A.; Bolognese, A.; Il Farmaco 1997, 52, 237.
- 6. Koike, H.; Imanashi, N.; Natsume Y.; Morooka, S.; Eur. J. Pharm. Mol. Pharm. 1994, 269;
- Tanabe, Y.; Yamamoto, H.; Murakami, M.; Yanagi, K.; Kubota, Y.; Okumura, H.; Sanemitsu, Y.; Suzukamo, G.; J. Chem. Soc. Perkin Trans. I 1995, 7, 935;
- Tanabe, Y.; Komuro, Y.; Imanishi, N.; Morooka, S.; Enomoto, M.; Kojima, A.; Sanemitsu, Y.; Mizutani, M.; Tetrahedron. Lett. 1991, 32 379.
- 7. Diurno, M. V.; Mazzoni, O.; Correale, G.; Monterry, I. G.; Il Farmaco 1999, 54, 579;
- Previtera, T.; Vigorita, M. G.; Bisila, M.; Orsini, F.; Benetolla, F.; Bombieri, G.; Eur. J. Med. Chem. 1994, 29, 317.
- 8. Desai, S. B.; Desai, P. B.; Desai, K. R.; Asian J. Chem. 1999, 2, 363;
- Sharma, R. C.; Kumar, D.; J. Indian Chem. Soc. 2000, 77, 492;
- Piscapo, E.; Diurno, M. V.; Gagliardi, R.; Mazzoni, O.; Boll. Soc. Ital. Biol. Sper. 1989, 65, 853.
- 9. Ueno, H.; Oe, T.; Snehiro, I.; Nakamura, S. US Patent 5594116, 1997, Chem. Abstr. 126 (1977) 157507p.
- 10. Ottaná, R.; Mazzon, E.; Dugo, L.; Monforte, F.; Maccari, R.; Sautebin, L.; De Luca, G.; Vigorita, M. G.; Alcaro, S.; Ortuso, F.; Eur. J. Pharmacol. 2002, 448, 71.
- 11. Hara, A.; Suzuki, T.; Hashizume, H.; Shishido, N.; Nakamura, M.; Ushikubi, F.; Abiko, Y.; Eur. J. Pharmacol. 1999, 385, 81;
- Kato, T.; Ozaki, T.; Tamura, K.; J. Med. Chem. 1999, 42, 3134.
- 12. Tanabe, Y.; Suzukamo, G.; Komuro, Y.; Imanishi, N.; Morooka, S.; Enomoto, M.; Kojima, A.; Sanemitsu, Y.; Mizutani, M.; Tetrahedron Lett. 1991, 32, 379.
- 13. Kato, T.; Ozaki, T.; Ohi, N. Tetrahedron: Asymmetry 1999, 10, 3963.
- 14. Adachi, Y.; Suzuki, Y.; Homma, N.; Fukazawa, M.; Tamura, K.; Nishie, I.; Kuromaru, O.; Eur. J. Pharmacol. 1999, 367, 267.
- 15. Ebeid, M, Y.; Fathallah, O, A.; El-Zaher, M. I.; Kamel, M, M.; Abdon, W, A.; Anwar, M. M.; Bull. Fac. Pharm. 1996, 34 125.
- 16. Rawal, R.K.; Prabhakar, Y.S.; Katti S.B.; De Clercq E.; Bioorg. Med. Chem. 2005, 13, 6771.
- 17. Kato, Y.; Kita, Y.; Nishio, M.; Hirasawa, Y.; Ito, K.; Yamanaka, T.; Motoyama, Y.; Seki, J.; Eur. J. Pharmacol. 1999, 384, 197.
- 18. Voss, M. E.; Carter, P.H.; Tebben, A. J.; Scherle, P. A.; Brown, G. D.; Thompson, L, A.; Xu, M.; Lo, Y, C.; Yang, Liu, R. R.-Q; Bioorg. Med. Chem. Lett. 2003, 13, 533.
- 19. Vicini, P.; Geronikaki, A.; Anastasia, K.; Incerti, M.; Zani, F.; Bioorg. Med. Chem. 2006, 14, 3859.
- 20. Blanchet, J.; Zhu J.; Tetrahedron Lett. 2004, 45, 4449.
- 21. Laurent, D. R. St.; Gao, Q.; Wu, D.; Serrano-Wu, M. H.; Tetrahedron Lett. 2004, 45, 1907.
- 22. Saeed, A.; Parvez, M.; Cent. Eur. J. Chem. 2005, 3, 780;
- Saeed A.; Flörke, U.; Acta Cryst. 2006, E62, o2403;
- Saeed, A.; Flörke, U.; Acta Cryst. 2006, E62, o2530.
- 23. Durust Y.; Nohut F.; Synth. Commun. 1999, 29, 1997.
- 25. Bruker (2002). SMART (Ver. 5.62), SAINT (Ver. 6.02), SHELXTL (Ver. 6.10) and SADABS (Version 2.03). Bruker AXS Inc., Madison, Wisconsin, USA.
- 26. Cappuccino, J.G.; Sherman, N. In Microbiology: a Laboratory Manual, pp. 247-251, The Benjamin/Cummings publishing Co.: California (USA) (1996).
Publication Dates
-
Publication in this collection
28 June 2007 -
Date of issue
2007
History
-
Received
23 Aug 2006 -
Accepted
27 Apr 2007