Abstracts
Eleven acridone alkaloids isolated from Swinglea glutinosa(Bl.) Merr. were examined for in vitroactivity against chloroquine-sensitive Plasmodium falciparum3D7, Trypanosoma brucei rhodesienseSTIB900 and Leishmania donovaniL82. An assay with KB cells was developed in order to compare in vitrotoxicity of alkaloids with the selective action on the parasites. Nine of the compounds had IC50 values ranging from 0.3 to 11.6 µM against P. falciparum. In contrast, a small number of compounds showed significant activity against T. brucei rhodesienseand none had activity against L. donovani. Among the alkaloids three had IC50 < 1.0 µM against P. falciparum, whereas against T. b. rhodesiensefive had IC50 < 10 µM. The characterization of the acridone alkaloids, 1,3,5-trihydroxy-4-methoxy-10-methyl-2,8-bis(3-methylbut-2-enyl)acridin-9(10H)-one (1), 2,3-dihydro-4,9-dihydroxy-2-(2-hydroxypropan-2-yl)-11-methoxy-10-methylfuro[3,2-b] acridin-5(10H)-one (2) and 3,4-dihydro-3,5,8-trihydroxy-6-methoxy-2,2,7-trimethyl-2Hpyrano[2,3-a]acridin-12(7H)-one (3), is discussed, as well as the structure-activity relationship of all compounds assayed. Isolation and spectral data of alkaloids 1-3are described for the first time although their citotoxicities to cancer cells have been described before.
antiparasitic acridone alkaloids; malaria; Swinglea glutinosa; Rutaceae
Onze alcalóides acridônicos isolados de Swinglea glutinosa(Bl.) Merr. foram avaliados para suas atividades in vitrocontra linhagens de Plasmodium falciparumsensíveis a cloroquina 3D7, Trypanosoma brucei rhodesienseSTIB9000 e Leishmania donovaniL82. Ensaios com células KB foram também executados com o objetivo de se determinar o grau de toxicidade das substâncias ativas contra os parasitas. Nove dos compostos apresentaram IC50 entre 0,3 e 11,6 µM contra P. falciparum. Em contraste, um pequeno número de compostos mostrou atividade significativa contra T. brucei rhodesiensee nenhum apresentou atividade contra L. donovani. Entre os alcalóides três tiveram IC50 < 1,0 µM contra P. falciparum, enquanto que contra T. b. rhodesiensecinco mostraram IC50 < 10 µM. A caracterização dos alcalóides, 1,3,5-triidróxi-4-metóxi-10-metil-2,8bis(3-metilbut-2-enil)acridin-9(10H)-ona (1), 2,3-diidro-4,9-diidróxi-2-(2-hidróxipropan-2-il)11-metóxi-10-metilfuro[3,2-b]acridin-5(10H)-ona (2) e 3,4-diidro-3,5,8-triidróxi-6-metóxi-2,2,7trimetil-2H-pirano[2,3-a]acridin-12(7H)-ona (3), é aqui discutida. Discute-se também a relação estrutura-atividade para todos os compostos ensaiados. O isolamento e dados espectrais para os alcalóides 1-3estão sendo aqui descritos pela primeira vez, embora em trabalho anterior tenham sido relatadas as suas atividades citotóxicas.
ARTICLE
Antiparasitic activities of acridone alkaloids from Swinglea glutinosa (Bl.) Merr.
Djalma A. P. dos SantosI; Paulo C. VieiraI, * * e-mail: paulo@dq.ufscar.br ; M. Fátima das G. F. da SilvaI; João B. FernandesI; Lauren RattrayII; Simon L. CroftII
IDepartamento de Química, Universidade Federal de São Carlos, CP 676, 13565-905 São Carlos-SP, Brazil
IIDepartment of Infectious and Tropical Diseases, London School of Hygiene & Tropical Medicine, London, WC1E 7HT, UK
ABSTRACT
Eleven acridone alkaloids isolated from Swinglea glutinosa(Bl.) Merr. were examined for in vitroactivity against chloroquine-sensitive Plasmodium falciparum3D7, Trypanosoma brucei rhodesienseSTIB900 and Leishmania donovaniL82. An assay with KB cells was developed in order to compare in vitrotoxicity of alkaloids with the selective action on the parasites. Nine of the compounds had IC50 values ranging from 0.3 to 11.6 µM against P. falciparum. In contrast, a small number of compounds showed significant activity against T. brucei rhodesienseand none had activity against L. donovani. Among the alkaloids three had IC50 < 1.0 µM against P. falciparum, whereas against T. b. rhodesiensefive had IC50 < 10 µM. The characterization of the acridone alkaloids, 1,3,5-trihydroxy-4-methoxy-10-methyl-2,8-bis(3-methylbut-2-enyl)acridin-9(10H)-one (1), 2,3-dihydro-4,9-dihydroxy-2-(2-hydroxypropan-2-yl)-11-methoxy-10-methylfuro[3,2-b] acridin-5(10H)-one (2) and 3,4-dihydro-3,5,8-trihydroxy-6-methoxy-2,2,7-trimethyl-2Hpyrano[2,3-a]acridin-12(7H)-one (3), is discussed, as well as the structure-activity relationship of all compounds assayed. Isolation and spectral data of alkaloids 1-3are described for the first time although their citotoxicities to cancer cells have been described before.
Keywords: antiparasitic acridone alkaloids, malaria,Swinglea glutinosa, Rutaceae
RESUMO
Onze alcalóides acridônicos isolados de Swinglea glutinosa(Bl.) Merr. foram avaliados para suas atividades in vitrocontra linhagens de Plasmodium falciparumsensíveis a cloroquina 3D7, Trypanosoma brucei rhodesienseSTIB9000 e Leishmania donovaniL82. Ensaios com células KB foram também executados com o objetivo de se determinar o grau de toxicidade das substâncias ativas contra os parasitas. Nove dos compostos apresentaram IC50 entre 0,3 e 11,6 µM contra P. falciparum. Em contraste, um pequeno número de compostos mostrou atividade significativa contra T. brucei rhodesiensee nenhum apresentou atividade contra L. donovani. Entre os alcalóides três tiveram IC50 < 1,0 µM contra P. falciparum, enquanto que contra T. b. rhodesiensecinco mostraram IC50 < 10 µM. A caracterização dos alcalóides, 1,3,5-triidróxi-4-metóxi-10-metil-2,8bis(3-metilbut-2-enil)acridin-9(10H)-ona (1), 2,3-diidro-4,9-diidróxi-2-(2-hidróxipropan-2-il)11-metóxi-10-metilfuro[3,2-b]acridin-5(10H)-ona (2) e 3,4-diidro-3,5,8-triidróxi-6-metóxi-2,2,7trimetil-2H-pirano[2,3-a]acridin-12(7H)-ona (3), é aqui discutida. Discute-se também a relação estrutura-atividade para todos os compostos ensaiados. O isolamento e dados espectrais para os alcalóides 1-3estão sendo aqui descritos pela primeira vez, embora em trabalho anterior tenham sido relatadas as suas atividades citotóxicas.
Introduction
Parasitic protozoa are the causative agents of human and livestock diseases infecting hundreds of millions of people every year and are collectively one of most important causes of human misery.1 Human African trypanosomiasis (HAT), or sleeping sickness, malaria, Chagas' disease and leishmaniasis are major health problems in many countries. HAT promoted by Trypanosoma brucei rhodesiense and T. b. gambiense is endemic in over 30 African countries threatening over 60 million people. HAT has reached epidemic proportions in some countries, such as Angola, southern Sudan, Uganda and the Democratic Republic of Congo.2,3 Malaria has re-emerged as a major public health problem over the past three decades mainly because of the development of worldwide resistance of Plasmodium falciparumto chloroquine, a drug which formed the basis for cheap and effective treatment and for prophylaxis of this disease.4 Each year, approximately 300 to 500 million malaria infections lead to over one million deaths. In many endemic countries, malaria is responsible for economic stagnation, lowering the annual economic growth in some regions by up to 1.5%.5,6 Leishmaniasis is a disease caused by protozoa of the genus Leishmania. According to WHO 88 countries are affected, with 350 million people at risk. 90% of cases of visceral leishmaniasis occur in India, Sudan, Bangladesh and Brazil.7 Present chemotherapy for these diseases is inadequate or toxic, or becoming ineffective due to an increase in resistance.8
The family Rutaceae contains many secondary metabolites such as alkaloids, flavonoids, coumarins, limonoids and lignans with a large spectrum of biological activities.9 Studies showed that acridone alkaloids are compounds with promising activity against P. falciparum,10,11 and also have antiviral12 and antiproliferative effects on cancer cell lines.13,14 The Asian genera Citrusand Swingleaare members of the Rutaceae and are included in the subfamily Aurantioideae. Citrusspecies have been investigated and characterized by possessing acridone alkaloids. These data stimulated an investigation of Swinglea glutinosa(Bl.) Merr. in a search for lead acridones.
From the MeOH extract of the stem bark of S. glutinosathree new acridone alkaloids were identified: 1,3,5-trihydroxy-4-methoxy-10-methyl-2,8-bis(3-methylbut-2-enyl)acridin-9(10H)-one (1), 2,3-dihydro4,9-dihydroxy-2-(2-hydroxypropan-2-yl)-11-methoxy-10-methylfuro[3,2-b]acridin-5(10H)-one (2) and 3,4-dihydro-3,5,8-trihydroxy-6-methoxy-2,2,7-trimethyl2H-pyrano[2,3-a]acridin-12(7H)-one (3). In addition, eight known alkaloids were characterized by the analysis of their NMR spectra and compared with reference data: 1,3,5-trihydroxy-2,8-bis(3-methylbut-2-enyl)-10-methyl9-acridone (4),10 glycocitrine-IV (5),15 citrusinine-II (6),16 citrusinine-I (7),17 citibrasine (8),16 5-hydroxynoracronycine (9),18 pyranofoline (10)19 and bis-5-hydroxynoracronycine (11)20 . These alkaloids were tested in vitroagainst chloroquine-sensitive P. falciparum3D7, T. b. rhodesienseSTIB900,L. donovaniL82 and their toxicity effects were also evaluated on KB cells.

Clique to extend

Results and Discussion
Compound 1was isolated as an amorphous powder, the molecular formula was determined as C25H29NO5by HRESIMS, showing an [M+H]+ peak at m/z424.2124. The UV absorptions bands at λ max/nm: 222, 265, 285, 332 and 410 nm, IR absorptions at ν max/cm-1: 3500 and 1625 cm and the characteristic signal of a hydrogen-bonded hydroxyl proton at d14.42 in the 1H NMR spectra, exchangeable with D2O, suggested the presence of a hydroxyl group. These data together with analyses of the 13C NMR spectrum, suggested that compound 1had a 1-hydroxy-9-acridone framework.16-22
The 1H NMR spectrum of the compound 1showed two AB type aromatic protons at δ 7.17 (1 H, d, J8.0Hz, H-6) and 6.96 (1H, d, J8.0Hz, H-7), one N-methyl group at δ 3.74 and one O-methyl group at δ 3.76. Also signals of two trisubstituted double bonds at δ 5.41(1H, m, H-2''), 5.31 (1H, m, H-2'), two methylene protons at δ 4.01 (2H, d, J7.0Hz, H-1''), 3.40 (2H, d, J7.0Hz, H-1'), the former protons being deshielded by the 9-carbonyl group, and four vinyl methyl groups [d 1.80 (3H,d, J0.9Hz, H-4' E), 1.65 (3H,d, J1.0Hz, H-5' Z), 1.74 (3H,d, J0.9Hz, H-4'' E), 1.69 (3H,d, J1.0Hz, H-5''Z)] were observed, which suggested the presence of two prenyl groups (3-methylbut-2-enyl). Their positions were confirmed at C-2 and C-8 by HMBC.
The position of a prenyl group at C-2 was confirmed by the correlation of the methylene protons at δ 3.40 (H-1') with C-1 (δ 158.1), C-2 (d 109.4), C-3 (d 156.1), C-2' (δ 123.6) and C-3' (δ 131.4) in the HMBC spectrum. In addition, the correlation of 1-OH at d 14.42 with C-1 and C-9a (δ 107.6) confirmed that the first prenyl group was attached to C-2. The position of the second prenyl group was confirmed by the correlation of the deshielded methylene protons at δ 4.01 (H-1'') with C-7 (δ 125.4), C-8 (δ 135.0), C-2'' (δ 125.4) and C-3'' (δ 131.2), confirming that the second prenyl group was attached to C-8. The N-methyl protons showed correlation with C-4a and C-5a at δ 140.2 and 139.4, respectively. The O-methyl protons showed correlation with C-4 at d 128.8. This value is consistent with the acridone possessing an O-methyl group at C-4.17,23 Based on these data, the structure of 1was characterized as 1,3,5-trihydroxy-4-methoxy-10methyl-2,8-bis(3-methylbut-2-enyl)acridin-9(10H)-one. Table 1 shows the data of 1H, 13C NMR and correlations observed in the HMBC experiment.
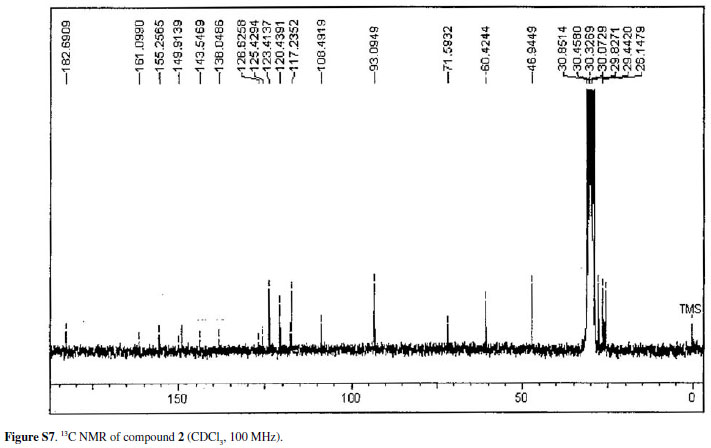
Compound 2was isolated as an amorphous powder, the molecular formula C20H21NO6was determined on the basis of HRESIMS, exhibiting an [M+H]+ peak at m/z372.1447. The UV and IR spectra were identical to those of 1, however some differences were observed in the 1H and 13C NMR. In the 1H NMR spectrum, the characteristic signal of a hydrogen-bonded hydroxy proton at δ14.42, exchangeable with D2O suggested the presence of a hydroxyl group. These data suggested that compound 2had a 4-hydroxy5-acridone skeleton.16-22
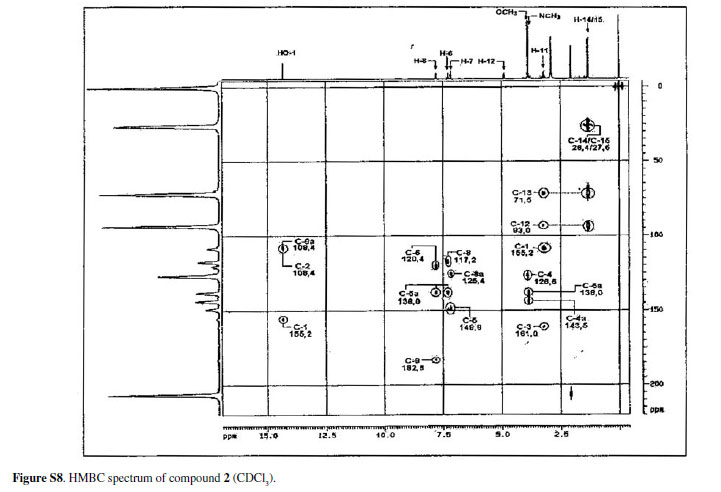
The 1H NMR spectrum of 2showed an ABX type aromatic spin system at δ 7.15 (1 H, t, J7.8Hz, H-7), 7.29 (1H, dd, J7.8, 1.4Hz, H-8) and 7.80 (1H, dd, J7.8, 1.4Hz, H-6), this last proton being deshielded by the 5-carbonyl group. The spectrum also showed one N-methyl group at d 3.85 and one O-methyl group at d 3.89. The presence of a hydroxyisopropyldihydrofuran moiety was suggested by an oxymethine proton at δ 4.88 (1H, dd, J9.4, 7.8Hz, H-2), methylene protons as two dd in an AB system at δ 3.20 (1H, dd, J15.5, 7.8Hz, H-3a) and 3.26 (1H, dd, J15.5, 9.4Hz, H-3b), two methyl groups at δ 1.33 (3H, s, H-2') and 1.29 (3H, s, H-3'). The signal at d 93.0 and 71.5 in the 13C NMR spectrum supported the presence of this substituent in the acridone nucleus.23
The presence of a linear orientation of the hydroxyisopropyldihydrofuran moiety was confirmed by correlations of the methylene protons at δ 3.20 (H-3) with C-3a (δ 109.4), C-2 (δ 93.0) and C-1' δ 71.5 and the correlations of the 4-OH at δ 14.31 with C-4a (δ 108.4), C-4 and C-3a in the HMBC spectrum. The spectrum also showed the correlation of the signals of the N-methyl protons at d 3.85 with d 143.5 and 138.0 assigned to C-11a and C-9a, respectively. The O-methyl proton at d 3.89 showed correlation with C-11 at δ 126.6. The correlation at H-6, H-7 and H-8 in the HMBC spectrum allowed the assignments of the carbons C-9, C-9a, C-8, C-7, C-6, C-5a and C-5. Based on the above evidence, compound 2could be defined as 2,3-dihydro-4,9-dihydroxy-2-(2hydroxypropan-2-yl)-11-methoxy-10-methylfuro[3,2-b] acridin-5(10H)-one. Table 2 shows 1H, 13C NMR data for 2 and the correlations observed in the HMBC.
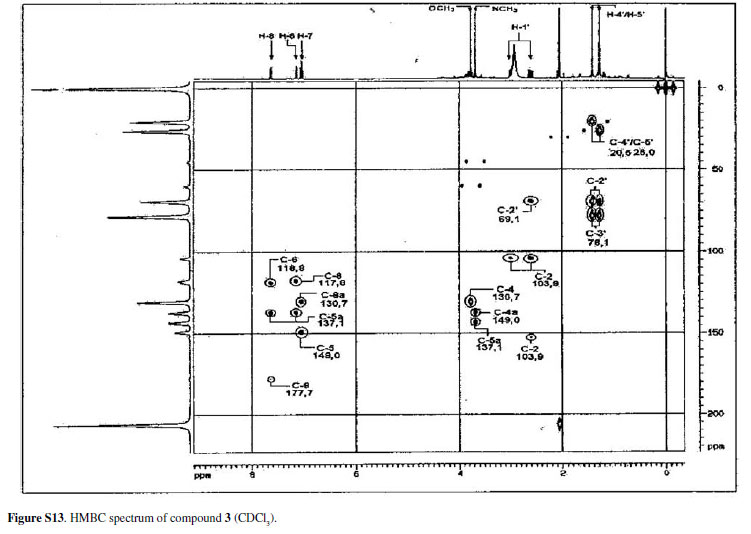
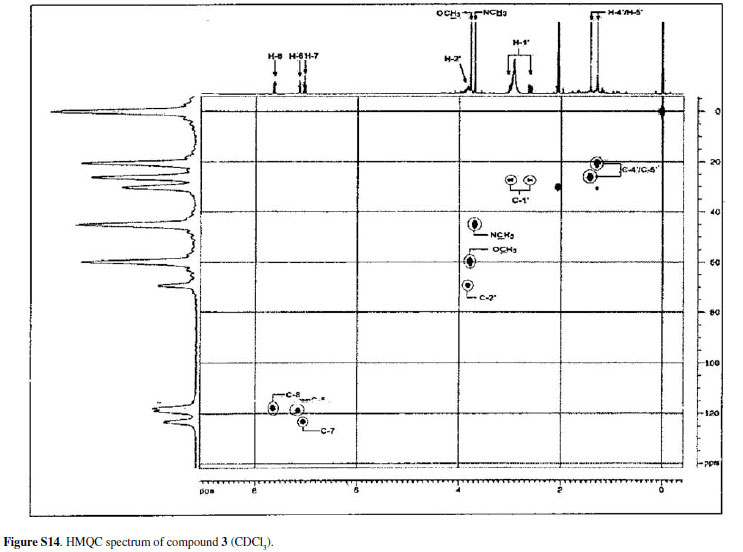
Compound 3was isolated as an amorphous powder, the molecular formula was determined as C20H21NO6 by HRESIMS, showing an [M + H]+ peak at m/z372.1447. The UV absorption band at λ max 265, 285, 332 and 410 nm, IR absorptions at ν max 3509 and 1620 cm-1 and comparison with 1H and 13C NMR data characterized an acridone nucleus.16-22 The lack of a characteristic signal of hydrogen-bonded hydroxy proton in the 1H NMR spectrum, in acetone-d6 and DMSO-d6, and the shielded value of C-12 at δ 177.7 in the 13C NMR spectrum suggested that there is no intramolecularly hydrogen-bonded hydroxyl group as observed for 1and 2. The 1H NMR spectrum showed an ABX type aromatic spin system at δ 7.04 (1H, t, J7.8Hz, H-10), 7.15 (1H, dd, J7.8, 1.5Hz, H-9) and 7.64 (1H, dd, J7.8, 1.5 Hz, H-11). The spectrum also showed one N-methyl group at δ 3.65 and one O-methyl group at δ 3.77. The presence of a 3-hydroxy-2,2-dimethyldihydropyran moiety was suggested by an oxymethine signal at δ 3.88 (1H, m, H-3), two methylene protons Ha at d 3.00 (1H, dd, J16.8, 5.7Hz, Ha-4), Hb 2.61 (1H, dd, J16.8, 7.8Hz, Hb-4) and two methyl groups at δ 1.43 (3H, s, H-1') and 1.28 (3H, s, H-2'). The resonances at δ 69.1 and 78.1 in the 13C NMR spectrum supported the presence of this substituent on the acridone nucleus.
In the HMBC spectrum of 3,the methylene protons at δ 2.61 (H-4) showed correlations with C-4a (δ 103.9), C-5 (δ 153.3), C-3 (δ 69.1) and C-2 (δ 78.1), indicating that the substituent was located at an angular position. This spectrum also showed the correlation of the N-methyl protons at d 3.65 with C-6a (δ 143.2) and C-7a (δ 137.1). The O-methyl protons at δ 3.77 showed correlation with C-6 at δ 130.4; this value is consistent with an acridone having an O-methyl group in this position.17,23 Correlations of H-9, H-10 and H-11 allowed the assignments of the carbons C-8, C-8a, C-9, C-10, C-11, C-11a and C-12. Therefore, compound 3was identified as 3,4-dihydro-3,5,8-trihydroxy-6-methoxy-2,2,7-trimethyl-2H-pyrano[2,3-a]acridin-12(7H)one. Table 3 shows the data of 1H, 13C NMR and the correlation observed in the HMBC.
The eleven acridone alkaloids isolated from S. glutinosawere tested for in vitroactivity against P. falciparum, T. b. rhodesienseand L. donovani. An assay with KB cells indicated in vitrocytotoxicity. The results are summarized in Table 4. To facilitate the discussion, IC50 values were assigned as IC50T,P,Kagainst T. b. rhodesiense, P. falciparum and KB cells, respectively.
Nine out of the eleven acridone alkaloids showed ICP50 below 10 µM, four showed ICT50 below 10 µM and none displayed significant activity against L. donovani.
Related acridone alkaloids from Thamnosma rodesica(Bak. F.), showed activity against promastigote and amastigote forms of Leishmania major.These alkaloids have a methyl 2,3-dihydroxypropanoate chain at C-3 and C-4,23 indicating that the substitution in the acridone skeleton is important for activity in this class of compounds.
According to the data, compound 5, with one prenyl group at C-2, was the most active against P. falciparum with ICP 0.3 µM. Comparison of 5, 1(ICP 2.6 µM) and 4 (ICP50 2.6 µM) indicates that the second prenyl at C-8 was responsible for reducing the activity of this series. This fact was also observed by Weniger et al.,10 who performed the assay with four alkaloids from S. glutinosaon a Nigerian chloroquine-sensitive strain of P. falciparum. Analysis of the results for compounds 2, 3, 9, 10and 11 suggests that the presence of a pyran ring is important for activity against P. falciparumand the position of this group, angular pyrano[2,3-c] (9) or linear pyrano[3,2-b] (10), did not alter the results of ICP50.
The activity against P. falciparumobserved for 6, 7and 8, ICP50 8.9, 29.9 and 6.1 µM, respectively, shows that presence of an O-methyl group at C-2 improves the activity.
From the 11 alkaloids tested against T. b. rhodesiense, compound 9 was the most active with an ICT50 1.0 µM.
Alkaloids 1-3had their citotoxicity to cancer cells described in an earlier paper,30 however here we disclose for the first time their isolation, spectral data and structure elucidation.
Experimental
General
Optical rotations were measured using a Perkin Elmer polarimeter. IR spectra were recorded on a Bomem M-B Series spectrophotometer. UV absorptions were recorded using a Varian 500 SCAN UV-Vis-NIR spectrophotometer. 1H and 13C NMR data were recorded on Bruker ARX-200 and Bruker DRX-400 spectrometers. Spectra were recorded in acetone-d6 and DMSO-d6 with TMS as internal standard. All 2D NMR data were recorded at 400MHz (Bruker DRX-400), HSQC J145 Hz; HMBC J8 Hz. HR-MS data were recorded on a Micromass Q-Tof (QqTOF) spectrometer; column chromatography was on silica gel 60 (Merck) and Sephadex LH-20 (Pharmacia). Preparative HPLC was performed on a Shodex Asahipak GS-310 2G column. TLC was carried out using Merck aluminum-backed silica gel 60 F254.
Plant material
Leaves, stem and root bark were collected in Campinas (SP) at the Instituto Agronômico de Campinas, and dried in the shade. The plant was identified by Prof. Dr. Maria Inês Salgado. A voucher specimen is deposited at the Herbarium of the Departamento de Botânica of the Universidade Federal de São Carlos (HUFSCar) as number 7110.
Extraction and isolation
The parts of the plant were extracted separately with n-hexane at room temperature for 3 days, filtered and evaporated under reduced pressure at 40 ºC. This procedure was repeated 3 times to yield the crude hexane extract. The residue was extracted, as above, using MeOH to yield the crude MeOH extract. The crude MeOH extract (60 g) was fractioned by VCC over 1 kg of silica gel 60 (70-239 mesh, Merck) eluted with 1.5 L of the solvents: 100% hexane (fraction 1), 100% CH2Cl2 (fraction 2), 3:1 CH2Cl2:MeOH (fraction 3); 2:1 CH2Cl2:MeOH (fraction 4); 1:1 CH2Cl2:MeOH (fraction 5) and 100% MeOH (fraction 5). Fractions 2 and 3 were combined and chromatographed on silica gel using CH2Cl2 as mobile phase, the polarity was increased by addition of 5%, 10%, 13%, 18%, 20% and 25% of MeOH in a gradient system to yield 15 fractions. Lupeol was crystallized from fractions 1 to 3. Fraction 5 was rechromatographed on Si gel, being eluted with 1:3 n-hexane-acetone followed by Sephadex LH-20 with MeOH to give alkaloids 1(16.3 mg), 4(30.4 mg), 5(3.5 mg), 8(11.2 mg), 9(13.4 mg), 10(9.8 mg) and 11 (5.4 mg). Further purification of fraction 9 on Sephadex LH-20 with MeOH and HPLC using MeOH as mobile phase, UV detection at 254 and 365nm, and a flow rate of 3.0 mL min-1 afforded compounds 6(13.5 mg), 7(11.2 mg) and 8(7.3 mg). Fraction 10 was dissolved in MeOH and submitted to Sephadex LH-20, being eluted with MeOH to give compounds 2(5.2 mg) and 3(4.3 mg).
1,3,5-Trihydroxy-4-methoxy-10-methyl-2,8-bis(3methylbut-2-enyl)acridin-9(10H)-one, (1)
Amorphous powder, IR (liquid film) ν max/cm-1: 3500, 2964, 1625, 1566. UV (MeOH) λ max/nm (log ε) 265 (3.47), 285 (3.08), 332 (3.02) and 410 (2.57). 1H, 13C NMR and HMBC correlations see Table 1. HRESIMS m/z 424.2124 [M + H]+ (calc. for C25H20NO5, 424.2124).
2,3-Dihydro-4,9-dihydroxy-2-(2-hydroxypropan-2-yl)-11methoxy-10-methylfuro[3,2-b]acridin-5(10H)-one, (2)
Optically inactive, amorphous powder, IR (liquid film) νmax /cm-1: 3475, 2964, 1630. UV (MeOH) λmax /nm (log ε): 259 (4.21), 274 (4.62), 284 (4.65). 1H, 13C NMR and HMBC correlations see Table 2. m/z372.1447 [M + H]+ (calc. for C20H22NO6, 372.1447).
3,4-Dihydro-3,5,8-trihydroxy-6-methoxy-2,2,7-trimethyl2H-pyrano[2,3-a]acridin-12 (7H)-one, (3)
[α]25D -21.4 (c, 0.0022 MeOH), amorphous powder, IR (liquid film) ν max/cm-1: 3509, 2978, 1620, 1572. UV (MeOH) λ max/nm (log ε): 264 (3.39), 285 (3.13), 332 (2.99). 1H, 13C NMR and HMBC correlations see Table 3. HRESIMS m/z372.1447 [M + H]+ (calc. for C20H22NO6, 372.1447).
Biological assays
Stock solutions of the compounds, plus control drugs, were prepared at a concentration of 20 mg mL-1 in DMSO (Sigma, UK), and diluted to appropriate concentrations prior to assays. IC50 values were calculated with MSXLFIT (IDBS, UK).
P. falciparum
Chloroquine-sensitive P. falciparumstrain 3D7 was maintained in human A+ erythocytes in RPMI 1640 medium (Sigma, UK) supplemented with Albumax II at 37 ºC in a 5% CO2-air mixture. Asynchronous (65-75% ring stage) of P. falciparumintraerythrocytic cultures were set up as above, with 1% parasitemia, 2.5% hematocrit, in triplicate in 100 µL of medium in 96 well, flat-bottomed Microtest III tissue plates. Drugs were added in a threefold dilution series and cultures incubated for a total of 48 h at 37 ºC in a 5% CO2-air mixture. After 24 h, {3H} hypoxanthine (0.2 mCi) was added to each well.25,26 At the end of the assay, plates were rapidly freeze-thawed, harvested using a Tomtec Mach III cell harvester (Tomtec, CT) onto a 96well format filtermat and MeltilexTM solid scintillant (both Wallac, Finland) added prior to reading in a Microbeta 1450 scintillation counter (Wallac, Finland) at 1 min per well.
T. brucei rhodesiense
T. b. rhodesienseSTIB900 bloodstream form trypomastigotes were maintained in HMI-18 medium,27 with 15% heat-inactivated fetal calf serum (Harlan Sera Lab, UK) at 37 ºC in a 5% CO2-air mixture. Prior to drugging, trypomastigotes were washed and resuspended in fresh medium at a concentration 2 × 105 trypanosoma/mL and 100 µL of this suspension was added to the drug dilutions. The top concentration for the test compounds was 30 mg mL-1. Pentamidine was included as the standard drug. Plates were incubated for 72 h at 37 ºC in a 5% CO2air mixture.28 At 72 h AlamarBlue was added to the plates. Plates were read after 4-5 h on a Gemini fluorescent plate reader (Sofimax Pro. 3.1.1, Molecular Devices, UK) at EX/EM 530/585 nm with a filter cut-off at 550 nm.
L. donovani
L. donovaniL82 amastigotes were harvested from an infected hamster (Mesocricetus auratus) spleen and used to infect murine peritoneal exudate macrophages (PEM) at a ratio of 7:1. In brief, infected cells were exposed to drug for a total of 5 days.29 The percentage of infected cells was evaluated microscopically and the percentage inhibition in comparison with untreated controls was calculated.
Cytotoxicity assays
96-well plates were seeded with KB cells at 4 × 104 mL-1 (100 µL perwell). Drugs at 300, 30, 3 and 0.3 µg mL-1 were added in fresh overlay after 24 h, in triplicate at each concentration. Plates were incubated for 72 h at 37 ºC in a 5% CO2-air mixture. At 72 h AlamarBlue was added to the plates. Plates were read after 4-5 h on a Gemini fluorescent plate reader (Sofimax Pro. 3.1.1, Molecular Devices, UK) at EX/EM 530/585 nm with a filter cut-off at 550 nm. IC50 values were calculated against the blanks and control samples.
Acknowledgments
The authors thank CNPq -Conselho Nacional de Desenvolvimento Científico e Tecnológico and Márcia Ortiz Mayo Marques from the Instituto Agronômico de Campinas. Lauren Rattray and Simon Croft received support from UNDP/World Bank/WHO Research Programme for Tropical Diseases (TDR).
Supplementary Information
Available free of charge at http://jbcs.org.br, as PDF file.
Received: November 27, 2008
Web Release Date: April 3, 2009
FAPESP helped in meeting the publication costs of this article.
Supplementary Information

Figure S1 - Click to enlarge

Figure S2 - Click to enlarge

Figure S3 - Click to enlarge

Figure S4 - Click to enlarge

Figure S5 - Click to enlarge

Figure S6 - Click to enlarge

Figure S7 - Click to enlarge

Figure S8 - Click to enlarge

Figure S9 - Click to enlarge

Figure S10 - Click to enlarge

Figure S11 - Click to enlarge

Figure S12 - Click to enlarge

Figure S13 - Click to enlarge

Figure S14 - Click to enlarge

Figure S15 - Click to enlarge
- 1. Barrett, M. P.; Burchmore, R. J. S., Stich, A.; Lazzari J. O.; Frasch, A. C.; Cazzulo, J.; Krishnas, J.; Lancet2003, 362, 1469.
- 2. Hoet, S.; Opperdoes, F.; Brun R.; Quetin-Leclercq, J. Q.; Nat. Prod. Rep 2004, 21, 353.
- 3. Legros, D.; Ollivier, G.; Gastellu-Etchegory, M.; Paquet, C.; Burri, C.; Jannin, J.; Buscher, P.; Lancet Infect. Dis.2002, 2, 437.
- 4. Wernsdorfer, W. H.; Acta Tropica1994, 56, 143.
- 5. Sachs, J.; Malaney, P.; Nature2002, 415, 680.
-
6WHO; www.rbm.who.int, accessed in February 18, 2004.
» link - 7. Sundar, S.; Trop. Med. Int. Health2001, 6, 849.
- 8. Croft, S. L.; Parasitology1997, 114, S3.
- 9. Da Silva, M. F. G. F.; Gottlieb, O. R.; Ehrendorfer, F.; Pl. Syst. Evol.1988, 161, 97.
- 10. Weniger, B.; Um, B-H.; Valentin, A.; Estrada, A.; Lobstein, A.; Anton, R.; Maillé, M.; Sauvain, M.; J. Nat. Prod 2001, 64, 1221.
- 11. Yamamoto, N.; Furukawa, H.; Ito, Y.; Yoshida, S.; Maeno, K.; Nishiyama, Y.; Antiviral Res 1989, 12, 21.
- 12. Kawaii, S.; Tomono, Y.; Katase, E.; Ogawa, K.; Yano, M.; Takemura,Y.;Motoharu, J.;Ito,C.;Furuhawa,H.; J. Nat. Prod 1999, 62, 587.
- 13. Chaya, N.; Terauchi, K.; Yamagata, Y.; Kinjo, J.; Okabe, H.; Biol. Pharm. Bull 2004, 27, 1312.
- 14. Wu, T-S.; Kuoh, C-S.; Furukawa, H.; Phytochemistry1983, 22,1493.
- 15. Ito,C.;Kondo,Y.;Wu,T-S.;Furukawa,H.; Chem. Pharm. Bull 2000, 48, 65.
- 16. Wu, T-S.; Furukawa, H.; Chem. Pharm. Bull 1983, 31, 901.
- 17. Furukawa, H.; Yogo, M.; Wu, T-S.; Chem. Pharm. Bull 1983, 31, 3084.
- 18. Wu, T-S.; Kuoh, C-S.; Furukawa, H.; Chem. Pharm. Bull 1983, 31, 895.
- 19. Wu, T-S.; Furukawa, H.; Kuoh, C-S.; Hsu, K-S.; J. Chem. Soc., Perkin Trans 11983, 1681.
- 20. Takemura, Y.; Wada, M.; Ju-ichi, M.; Ito, C., Furukawa, H.; Chem. Pharm. Bull 1998, 46, 693.
- 21. Pegel, K. H.; Wright, W.G.; J. Chem. Soc (C)1969, 2327.
- 22. Fraser, A. W.; Lewis, J.R.; J. Chem. Soc., Perkin Trans. 11973, 1173.
- 23. Wu, T-S.; Chen, C-M.; Chem. Pharm. Bull 2000,48, 85.
- 24. Ahua, K. M.; Ioset. J-R.; Ransijn, A.; Mauël, J.; Mavi, S.; Hostettmann, K.; Phytochemistry2004, 65, 968.
- 25. Desjardins, R. E.; Canfield, C. J.; Haynes, J. D.; Chulay, J.D.; Antimicrob. Agents Chemother.1979, 16, 710.
- 26. O'Neill, M. J.; Bray, D. H.; Boardman, P.; Phillipson, J. D.; Warhurst, D.C.; Planta Med.1985,61, 394.
- 27. Hirumi, H.; Hirumi, K.; J. Parasitol 1989, 75, 985.
- 28. Räz, B.; Iten, M.; Grether-Buler, Y.; Kamisky, R.; Brun, R.; Acta Tropica1997, 68, 139.
- 29. Neal, R.A.; Croft, S.L.; J Antimicrob. Chemother 1984, 14, 463.
- 30. Braga, P. A. C.; dos Santos, D. A. P.; da Silva, M. F. D. G. F.; Vieira, P. C.; Fernandes, J. B.; Houghton, P.J.; Fang, R.; Nat. Prod. Res.2007, 21, 47.
Publication Dates
-
Publication in this collection
10 June 2009 -
Date of issue
2009
History
-
Accepted
03 Apr 2009 -
Received
27 Nov 2008