Abstracts
Two new eremophilane sesquiterpenes, cupressolide A and cupressolide B, along with two known sesquiterpenes, has been characterized from the EtOAc extract of a liquid medium where a Xylariaceous fungus, isolated as an endophytic fungus from health tissues of Cupressus lusitanica leaves, was cultivated. The structures of the isolated compounds were determined by analyses of their MS and NMR spectroscopic data.
Cupressus lusitanica; Xylaria; eremophilane sesquiterpenes; endophytic fungi
Dois novos sesquiterpenos eremofilanos, cupressolideo A e cupressolideo B, além de dois outros conhecidos, foram isolados a partir do extrato AcOEt do meio de cultura de uma espécie de Xylaria, isolada como fungo endofítico dos tecidos sadios das folhas de Cupressus lusitanica. Estudos espectroscópicos, usando EM e RMN, levaram às estruturas dos dois sesquiterpenos de esqueleto eremofilanos, novos na literatura.
ARTICLE
Two novel eremophilane sesquiterpenes from an endophytic Xylariaceous fungus isolated from leaves of Cupressus lusitanica
Luciana S. Amaral; Edson Rodrigues-Filho* * e-mail: edson@dq.ufscar.br
Departamento de Química, Universidade Federal de São Carlos, CP 676, 13565-905 São Carlos-SP, Brazil
ABSTRACT
Two new eremophilane sesquiterpenes, cupressolide A and cupressolide B, along with two known sesquiterpenes, has been characterized from the EtOAc extract of a liquid medium where a Xylariaceous fungus, isolated as an endophytic fungus from health tissues of Cupressus lusitanica leaves, was cultivated. The structures of the isolated compounds were determined by analyses of their MS and NMR spectroscopic data.
Keywords:Cupressus lusitanica, Xylaria, eremophilane sesquiterpenes, endophytic fungi
RESUMO
Dois novos sesquiterpenos eremofilanos, cupressolideo A e cupressolideo B, além de dois outros conhecidos, foram isolados a partir do extrato AcOEt do meio de cultura de uma espécie de Xylaria, isolada como fungo endofítico dos tecidos sadios das folhas de Cupressus lusitanica. Estudos espectroscópicos, usando EM e RMN, levaram às estruturas dos dois sesquiterpenos de esqueleto eremofilanos, novos na literatura.
Introduction
Cupressus lusitanica, commonly known as a Mexican Cypress and Portuguese Cypress, belongs to the family Cupressaceae and is usually cultivated as an ornamental tree and in commercial forestry plantation.1,2 Due to its economic importance, this plant was included in our continuous program established to study the chemistry and biochemistry aspects of plant microorganisms interactions, with emphasis on those apparently symbiotic associations.
Among the endophytic fungi isolated from healthy tissues of C. lusitanica leaves, we obtained some strains with macro and micro morphology characteristics of those microorganisms belonging to the genus Xylaria. Besides these morphologic characteristics, Xylaria species are producers of secondary metabolites, including isocoumarin,3,4 cytochalasins,4-6 xanthones,7 xyloketals,8-10 sesquiterpenes,11 that contribute to their classification in this genus. In our study we detected isocoumarins and cytochalasins in the fungus extracts using mass spectrometry, which reinforce the hypothesis of its classification as a Xylareaceous fungus. Among the compounds isolated from the fungus extract, two novel (1 and 2) and two known (3 and 4) eremophilane sesquiterpenes were identified. The production of these terpenoids corroborated to classify the fungus within Xylariaceae family. Although it is not clear the importance of these compounds as phytotoxic agents, some members of this class of substances have shown remarkable biological activities, such as anti-inflammatory, antihyperglycemic, cytotoxic, HIV-1 integrase inhibitory.12-15
Results and Discussion
The EtOAc extract of liquid medium from endophytic fungi was chromatographed on silica gel columns to give four compounds (1-4). Compounds 3 and 4 were previously reported in the literature.16,17
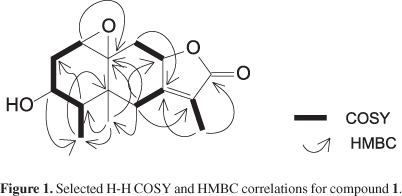
Compound 1 was obtained as a colorless crystal. The IR spectrum displayed a broad band at 3487 cm-1 characteristic of a hydroxyl group and a band at 1745 cm-1 attributed to a conjugated γ-lactone. The 13C NMR spectrum exhibited 15 signals which were assigned, by DEPT 135 and HSQC experiments to three methyls, three methylenes, four methines and three sp2 carbons, one of this being a carbonyl group. Its ESI-MS spectrum contains an ion peak of [M+H]+ at m/z 265, consistent with the molecular formula C15H20O4 which also was in accordance with the NMR data. The 1H NMR spectrum of 1 showed three signals δH 1.85, 0.83 and 0.96 attributed to CH3-13, CH3-14 and CH3-15, respectively. 1H NMR spectrum exhibited the presence of a deshielded signal assignable to an oxymethine hydrogen at δH 3.48 (1H, m, H-3); this signal correlated in COSY spectrum with the δH 1.89 (1H, m, H-2b); 2.44 (1H, ddd, J 15.2; 8.4; 4.4 Hz, H-2a); 1.81 (1H, m, H-4). H-4 showed coupling with the methyl group at δH 0.96 (3H, J 6.8 Hz, CH3-15). The presence of an epoxy group was confirmed by the chemical shifts of H-1 (δ 3.06), C-1(δ 58.5), and C-10 (δ 63.2).
The COSY spectrum exhibited correlation peaks among the oxymethine hydrogen at δH 4.9 (1H, m, H-8) with hydrogens at δH 2.05 (1H, m, H-9a); 1.82 (1H, m, H-9b); 1.85 (3H, t, J 1.2 Hz, CH3-13).
HSQC analysis indicated the presence of one tetrasubstituted double bond which was associated with the carbons at δC 159.5; 122.3 (C-7 and C-11, respectively). On the other hand another double bond was observed in the 13C NMR spectrum, with characteristic chemical shifts of a carbonyl group at δC 174.6 attributed to C-12.
The HSQC spectrum exhibited correlations among the diasterotopic hydrogens at δH 2.23 (1H, br d, J 13.6 Hz, H-6b) and δH 2.76 (1H, d, J 13.6 Hz, H-6b) with the carbon at δC 35.1. The HMBC spectrum showed coupling of H-6b with C-5, C-7, C-8, C-10, C-11 and CH3-14. The COSY and HMBC analysis of 1 led to a partial structure as shown in Figure 1.
The relative stereochemistry of 1 was elucidated using nOe and COSY spectroscopy. The β orientation of H-3 was inferred from the nOe correlation with CH3-14 and CH3-15. H-8 (δ 4.90) should be α oriented as indicated by the homoallylic coupling with the methyl (CH3-13, δ 1.85) bounded to C-11 observed in the COSY experiment. This homoallylic coupling requires a 90º dihedral angle of the methyl group at C-11 with H-6α and H-8, witch was in agreement with the nOe experiment. Furthermore, the nOe spectrum showed correlations of H-6α with H-4 (δ 1.81) and of H-6β with CH3-13, CH3-14 and CH3-15. All NMR data are shown in the Table 1.
Compound 2 showed spectroscopic data very similar to those of 1, indicating the presence of an eremophilane skeleton. The IR spectrum displayed a broad band at 3508 cm-1 characteristic of a hydroxyl group. The 1H NMR experiment exhibited two olefinic hydrogens, three carbinolic hydrogens and two methyl groups. The EI-MS spectrum of 2 contains an ion peak of M+ at m/z 252, consistent with the molecular formula C15H24O3 and the data observed in the NMR spectrum. The 1H NMR spectrum of compound 2 exhibited deshielded signals due the presence of olefinic hydrogens at δH 5.26 (1H, d, J 0.8 Hz, H-12a) and δH 5.12 (1H, d, J 0.8 Hz H-12b). The COSY spectrum showed the coupling of H-12a with the carbinolic hydrogens at δH 4.19 (1H, dd, J 11.2; 0.8 Hz, H-13a) and δH 4.16 (1H, dd, J 11.2; 0.8 Hz, H-13b). The peak attributed to H-8 at δH 3.88 (1H, ddd, J 4.4; 10.0; 15.6 Hz) exhibited correlations in the COSY spectrum with the hydrogens at δH 2.43 (1H, ddd, J 4.4; 8.0; 13.2 Hz, H-7), δH 2.18 (1H, dd, J 10.0; 12.4 H-9a) and δH 1.38 (1H, m, H-9b).
The presence of a methine group at δH 1.71 (1H, m, H-4) was confirmed by COSY correlations at δH 1.21 (1H, m, H-3a) and δH 1.71 (1H, m, H-3b) and δH 0.71 (3H, d, J 5.6 Hz, CH3-15). The signal of CH2-13 (δ 4.16 and 4.19), CH3-14 (δ 1.02), CH3-15 (δ 0.71) and CH2-12 (δ 5.12 and 5.26) in the 1H NMR spectrum were in agreement with the profile of an eremophilane with a double bound at C-11 and C-12. The epoxide group at C-1 and C-10 was deduced from the chemical shifts of H-1 (δ 2.97), C-1 (δ 59.7), and C-10 (δ 65.8). The COSY analysis of 2 led to a partial structure as shown by bold-faced lines in Figure 2, which were supported by HMBC correlations (Table 2). The relative stereochemistry was based on those determined to compound 2. All NMR data can be observed in Table 2.
Conclusions
The genus Xylaria is known for being a rich source of structurally diverse natural products including isocoumarin,3,4 cytochalsins,4-6 xyloketals,8-10 sesquiterpenes11 and others. Among these compounds eremophilane sesquiterpenes stand out for several biological activities.12-15 One member of this genus reported in the present study showed the notable ability to produce eremophilane sesquiterpenes, including two new in the literature, cupressolide A and cupressolide B. Due to the many biological activities shown by this class of compounds, this Xylaria deserves a careful study aiming to sesquiterpene production enhancement.

Experimental
Equipment
IR spectra were run on a Bomen MB102 -IR spectrometer using KBr pellets. Optical rotation was measured on a Perkin-Elmer 241 polarimeter. GC/MS analyses were performed using GC 8000 series Fisons and VG Platform mass spectrometer detector. 1D and 2D NMR spectra were obtained in CDCl3 (Aldrich) on DRX 400 Bruker spectrometer operating at 400 MHz for hydrogen and 100 MHz for 13C and TMS was used as internal standard. MS were acquired in positive ion mode on a triple quadrupole Micromass Quattro LC spectrometer, equipped with an ESI ion source.
Plant material
Health leaves of Cupressus lusitanica were collected in São Carlos, São Paulo State, Brazil. A voucher specimen (No. 7281) has been deposited in the Herbarium of the Botanic Department of Universidade Federal de São Carlos, Brazil.
Fungal material
The method of surface sterilization employed in this work was similar to that used by Petrini et al.18 After the collection, the leaves were washed in abundant water (domestic use grade) and then in distilled water. The leaves were surface sterilization by consecutive immersion in 70% ethanol (2 s), sterile distilled water (2 s), 11% aqueous sodium hypochloride for 1-5 min and 70% ethanol (2 s), and then in sterile distilled water. The material was placed in Petri dishes containing PDA medium (potato, dextrose and agar) supplemented with 100 μg mL-1 terramicin and incubated at room temperature. Endophytic fungi growing from the plant tissues, were picked and recultured on PDA to determine culture purity. It was deposited at LaBioMMi - Laboratório de Bioquímica Micromolecular de Microorganismos - of the Departamento de Química at Universidade Federal de São Carlos, Brazil. Working stocks were prepared on potato dextrose agar.
Fermentation and extraction
The fungus was grown under static conditions at room temperature for 20 days in 20 Erlenmeyer flasks containing the liquid medium (300 mL per flask) composed of glucose (26.7 g L-1), yeast extract (10.0 g L-1 ), NaNO3 (3.0 g L-1), K2HPO4 (1.0 g L-1), MgSO47H2 O (0.5 g L-1), KCl (0.5 g L-1), FeSO47H2 O (0.01 g L-1). The mycelium was separated by reduced pressure filtration and the liquid phase was partitioned with ethyl acetate (1,000 mL × 3). The organic solvent was dried with anydrous sodium sulfate, filtered and removed using vaccum to give the crude extract. Crude extract was analyzed by GC/MS. This technique enabled the detection of the eremophilane sesquiterpene valencene (4). The extract was chromatographed on silica gel columm (h = 4.5 cm and ∅ = 3.7 cm) eluted with a Hex:CH2Cl2 (1:1); CH2Cl2:EtOAc (1:1); CH2Cl2:EtOAc (1:4); CH2Cl2:EtOAc:MeOH (1:4:10%); EtOAc:MeOH (1:1); MeOH (100%) to afford six fractions (A-F). The fractions C and D were rechromatographed on silica gel columm (h = 16 cm and ∅ = 2.5 cm) with Hexane 100% gradient to EtOAc:MeOH (1:1) to give 1 (5.2 mg), 2 (4.8 mg), 3 (2.7 mg).
GC/MS analysis
The extract was submitted to clean-up procedures using solid phase extraction (SPE). The SPE cartridge was activated with 100% Hexane and conditioned with 3 mL of CHCl3. The extract (10 mg) was dissolved in 3 mL of CHCl3 and loaded to the SPE cartridge. Elution of SPE cartridge with CHCl3 produced an apolar fraction. For the GC/MS analysis the injector temperature was kept at 180 ºC and the GC oven temperature was maintained at 70 ºC during 6 min and then increased to 250 ºC at a rate of 6 ºC min-1 and finally increased to 325 ºC at 3 ºC min-1. The sample volume injected was 2 μL.
MS data collection
ESI-MS data were colleted from direct introduction of the sample solution 5 μL of compound 1 (5 μg mL-1). The optimal voltages found for the probe and ion source components to produce maximum intensity of the ions [M+H]+ were 3.3 kV for the capillary, 19 V for the sample cone, and 4 V for the extractor cone.
Physical and spectral data
(1αR,3R,4R,4αR,8αR,9α S)-3-Hydroxy-4,4α,6-trimethyl-1 α,2,3,4,4αR,8α,9-octahydro-7H-oxirene[8,8 α] naphtho[2,3β] furan-7-one, compound 1: Cupressolide A
Colorless crystals (CH2Cl2); [α]D25 = -6.262 (c 0.0001, CHCl3); IR(KBr) νmax/cm-1: 3487, 1745; ESI-MS m/z 265 [M+H]+; ESI-MS-MS (20 eV) m/z 265 (78%), 247 (17), 229 (39), 201 (50), 183 (100), 173 (70), 147 (60), 119 (55); NMR data see Table 1.
(1αR,4S,4αR,6S,7R,8 αS)-6-[1-(Hydroxymethyl)vinyl]-4,4α-dimethyloctahydro-1α H-naphtho[1, 8αβ]oxiren-7-ol, compound 2: Cupressolide B
Colorless crystal (CH2Cl2); IR(KBr) νmax/cm-1: 3508; EIMS (70 eV) m/z 252 [M]+ (3%), 234 (6), 216 (12), 201 (16), 168 (57), 153 (75), 125 (100); NMR data see Table 2.
Supplementary Information
Supplementary data are available free of charge at http://jbcs.sbq.org.br, as PDF file.
Acknowledgments
The autors are gratefull to the Brazilian institutions FAPESP - Fundação de Amparo à Pesquisa do Estado de São Paulo, CNPq - Conselho Nacional de Desenvolvimento Científico e Tecnológico, CAPES - Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior for the financial support.
Received: June 25, 2009
Web Release Date: March 25, 2010
FAPESP has sponsored the publication of this article.
Supplementary Information











- 1. Farjon, A.; Taxon 1993, 42, 81.
- 2. Graniti, A.; Annu. Rev. Phytopathol. 1998, 36, 91.
- 3. Tansuwan, S.; Pornpakakul, S.; Roengsumran, S.; Petsom, A.; Muangsin, N.; Sihanonta, P.; Chaichit N.; J. Nat. Prod. 2007, 10, 1620.
- 4. Pongcharoen, W.; Rukachaisrikul, V.; Isaka, M.; Sriklung, K.; Chem. Pharm. Bull. 2007, 55, 1647.
- 5. Espada, A.; Rivera-Sagredo, A.; de la Fuente, J. M.; Hueso-Rodríguez, J. A.; Élson, S. W.; Tetrahedron 1997, 53, 6485.
- 6. Abate, D.; Abraham, W.; Meyer, H.; Phytochemistry 1997, 44, 1443.
- 7. Healy, P. C.; Hocking, A.; Tran-Dinh, N.; Pitt, J. I.; Shivas, R. G.; Mitchell, J. K.; Kotiw, M.; Davis, R. A.; Phytochemistry 2004, 65, 2373.
- 8. Lin, Y.; Wu,X.; Feng, S.; Jiang, G.; Luo, J.; Zhou, S.; Vrijmoed, L. L. P.; Jones, E. B. G.; Krohn, K.; Steingröver, K.; Zsila, F.; J. Org. Chem. 2001, 66, 6252.
- 9. Xiaobo, Z.; Haiying, W.; Linyu, H.; Yongcheng, L.; Zhongtao, L.; Process Biochem. 2006, 41, 293.
- 10. Liu, X.; Xu, F.; Zhang, Y.; Liu, L.; Huang, H.; Cai, X.; Lin, Y.; Chanb W.; Russ. Chem. Bull. 2006, 55, 1091.
- 11. McDonald, L. A.; Barbieri, L. R.; Bernan, V. S.; Janso, J.; Lassota, P.; Carter G. T.; J. Nat. Prod. 2004, 67, 1565.
- 12. Wang, W.; Gao, K.; Jia, Z.; J. Nat. Prod. 2002, 65, 714.
- 13. Garduño-Ramírez, M. L.; Trejo, A.; Navarro, V.; Bye, R.; Linares, E.; Delgado, G.; J Nat. Prod. 2001, 64, 432.
- 14. Li, E.-W.; Pan, J.; Gao, K.; Jia, Z.-J.; Planta Med. 2005, 71, 1140.
- 15. Zhang, Q. J.; Dou, H.; Zheng, Q. X.; Zhou, C. H.; Xu, Z. J.; Peng, H.; Zhao, Y.; Chin. Chem. Lett. 2005, 16, 360.
- 16. Zhao Y.; Schenk, D. J.; Takahashi, S.; Chappell, J.; Coates, R. M.; J. Org. Chem. 2004, 69, 7428.
- 17. Prez-Castorena, A. L.; Arciniegas, A.; Guzmn, S. L.; Villaseor, J. L.; de Vivar, A. R.; J. Nat. Prod. 2006, 69, 1471.
- 18. Petrini, O.; Sieber, T. N.; Toti, L.; Viret, O.; Natural Toxins 1992, 1, 185
Publication Dates
-
Publication in this collection
14 Oct 2011 -
Date of issue
2010
History
-
Received
25 June 2009