Abstracts
Pd/C facilitated dual C-C bond forming reaction between 2,4-diiodoquinoline and terminal alkynes in water providing a practical and one-step synthesis of 2,4-dialkynylquinolines. A number of related quinoline derivatives were prepared in good to excellent yields using this water-based methodology. The use of other palladium catalysts and solvents was examined and the mechanism of the reaction has been discussed.
quinoline; palladium; charcoal; copper; C-C bond; alkyne
Pd/C catalisa a reação de acoplamento entre 2,4-diiodoquinolina e alquinos terminais em água, proporcionando uma síntese prática, de uma etapa, de 2,4-dialquinilquinolinas. Uma série de derivados de quinolinas relacionadas foi preparada em bons a excelentes rendimentos, usando esta metodologia baseada em água. O uso de outros catalisadores de paládio e solventes foi examinado e o mecanismo de reação discutido.
ARTICLE
Pd/C-mediated dual C-C bond forming reaction in water: synthesis of 2,4-dialkynylquinoline
Ellanki A. ReddyI,II; Aminul IslamI; K. MukkantiII; Bolla K. VenuI; Manojit PalIII,* * e-mail: manojitpal@rediffmail.com
IDr. Reddy's Laboratories Limited, Bollaram Road, Miyapur, Hyderabad, 500049 Andhra Pradesh, India
IIChemistry Division, Institute of Science and Technology, JNT University, Kukutpally, Hyderabad, 500072 Andhra Pradesh, India
IIINew Drug Discovery, R&D Center, Matrix Laboratories Ltd., Anrich Industrial Estate, Bollaram, Jinnaram Mandal, Medak District, 502 325 Andra Pradesh, India
ABSTRACT
Pd/C facilitated dual C-C bond forming reaction between 2,4-diiodoquinoline and terminal alkynes in water providing a practical and one-step synthesis of 2,4-dialkynylquinolines. A number of related quinoline derivatives were prepared in good to excellent yields using this water-based methodology. The use of other palladium catalysts and solvents was examined and the mechanism of the reaction has been discussed.
Keywords: quinoline, palladium, charcoal, copper, C-C bond, alkyne
RESUMO
Pd/C catalisa a reação de acoplamento entre 2,4-diiodoquinolina e alquinos terminais em água, proporcionando uma síntese prática, de uma etapa, de 2,4-dialquinilquinolinas. Uma série de derivados de quinolinas relacionadas foi preparada em bons a excelentes rendimentos, usando esta metodologia baseada em água. O uso de outros catalisadores de paládio e solventes foi examinado e o mecanismo de reação discutido.
Introduction
The quinoline core (p-electron deficient heterocycle) being one of the privileged structures for the design and development of potential new drugs have found wide applications in pharmaceutical research.1-4 Though a wide variety of quinoline derivatives have been reported earlier a thorough literature search however revealed that 2,4-dialkynylquinolines surprisingly remained unexplored so far. As part of our ongoing program on building of quinoline-based library of small molecules we were in need of a wide range of 2,4-dialkynylquinolines and a convenient synthetic methodology to access these derivatives.
The palladium mediated alkynylation of aryl/heteroaryl halides under Sonogashira condition5 has become a highly effective tool for the introduction of an alkynyl moiety to aryl or heteroaryl ring.6 The reactivity of 2,4-dihaloquinolines towards alkynylation reaction under Sonogashira or modified conditions has been studied earlier (Scheme 1). For example 2,4-dibromoquinoline derivative provided the 2-alkynylated product A (Scheme 1) selectively when treated with a terminal alkyne.7 Similarly, 2,4-dichloroquinoline afforded a 2-alkynyl derivative when coupled with a terminal alkyne under Pd/C-Cu catalysis in water.8 Coupling of 2-bromo-4-iodoquinoline derivative with a terminal alkyne in the presence of (PPh3)2PdCl2-CuI though provided a 2,4-dialkynylated product B in 17% yield (Scheme 1) the 4-alkynyl derivative however was isolated as a major product in this case.7 Nevertheless, to the best of our knowledge this is the only example known so far for the preparation of 2,4-dialkynylquinoline (B) and no detailed study on this reaction has been reported. Therefore, the development of a general method leading to compound B (Z = H) exclusively was required. Herein, we report a direct and practical method for the preparation of 2,4-dialkynylquinoline under Pd/C-Cu catalysis in water. To the best of our knowledge this is the first example of Pd/C-mediated dual C-C bond forming reaction in water.
Results and Discussion
Initially, we attempted to prepare the 2,4-dialkynylquinoline from its 2,4-dichloro precursor via a two-step method. Thus 2-alkynyl quinoline prepared via earlier method8,9 was treated with a terminal alkyne in the presence of a number of palladium catalysts e.g. (PPh3)2PdCl2, Pd(OAc)2 or Pd/C. However, none of these reactions provided the desired product in good yields. It is well known that the chloro group is less reactive than iodo group towards palladium catalysts under normal Sonogashira conditions. Hence we decided to use a 2,4-diiodoquinoline (2) for our synthesis which could be readily obtained from the corresponding 2,4-dichloro analogue (1) as shown in Scheme 2.10
Having prepared the 2,4-diiodoquinoline (2) we initially coupled it with 1-octyne in the presence of various palladium catalysts (Table 1). Because of our earlier success in the use of 10% Pd/C-PPh3-CuI as a catalyst system8 in water we decided to conduct the reaction of 2 (1.0 equiv.) with 1-octyne (3.0 equiv.) in the presence of same catalysts in water using Et3N as a base. The reaction proceeded smoothly affording the desired product 3a in 85% yield (Entry 1, Table 1) and no monoalkynylated or other side product was detected in the reaction mixture. The reaction was carried out for 10 h and an increase of reaction time did not improve the product yield (Entry 2, Table 1). The product formation was almost suppressed in the absence of PPh3 (Entry 3, Table 1). The use of other catalysts, e.g. PdCl2(PPh3)2, Pd(PPh3)4 or Pd(OAc)2-PPh3 afforded the product 3a albeit in inferior yield (Entries 4-6, Table 1). While the use of other solvents, e.g. 1,4-dioxane, DMF and EtOH provided 3a in good yield (Entries 7-9, Table 1), water was however the solvent of our choice.
We then decided to explore the scope and generality of this Pd/C mediated dual C-C bond forming reaction on a quinoline ring in water. Thus a variety of terminal alkynes were reacted with 2 in the presence of 10% Pd/C, PPh3 and CuI in water (Scheme 2). The results of this study are summarized in Table 2. Terminal alkynes containing a range of groups in their side chain such as alkyl (Entries 1-3, Table 2), aryl (Entries 4-6, Table 2), cyano (Entry 7, Table 2) or hydroxyl alkyl moieties (Entries 8-10, Table 2) were reacted with the diiodo compound 2 and all these groups were well tolerated under the condition employed. The reaction proceeded well in water in all these cases affording the corresponding 2,4-dialkynylquinolines (3a-j) in good to excellent yields. All the products isolated were well characterized by spectral (NMR, IR and MS) and analytical data. The presence of alkyne moieties was indicated by a sharp IR absorption shown by all the quinoline derivatives synthesized in the region 2200-2230 cm-1. This was further supported by the appearance of signals corresponding to the sp-carbons at δ 70-96 in 13C NMR spectra of compounds 3a-j.
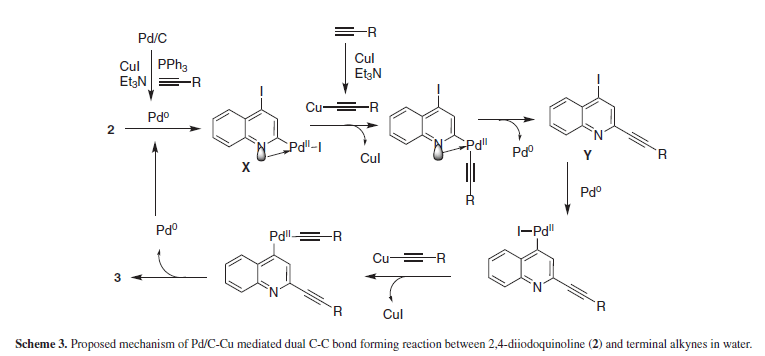
Mechanistically, the reaction may appeared to proceed via a simultaneous C-C bond forming reaction at C-2 and C-4 of the quinoline ring as neither C-2 nor C-4 mono alkynyl derivative was isolated as a side product from the reaction mixture. However, such a reaction would mechanistically be difficult as it would involve third-order kinetics in the Pd-mediated C-I oxidative addition and/or transmetallation process. Moreover, because of the use of elevated reaction temperatures and the fact that theoretically, the second alkynylation could be kinetically more facile than the first one; the real step-by-step process was practically not observed during the reaction. Nevertheless, because of high reactivity of the actual catalytic species Pd(0) generated11 in situ towards Csp2-I bond the organo-palladium intermediate (X) formed as a result of oxidative addition of Pd(0) to 2,4-diiodoquinoline would contain one Csp2-Pd-I moiety initially (Scheme 3). It is likely that the oxidative addition of Pd(0) would take place initially at C-2 of the quinoline ring8,9 due to the higher reactivity of iodo group at this position. The copper acetylide generated from the terminal alkyne and CuI would undergo trans metalation with the organo-palladium species and subsequent reductive elimination of Pd(0) would produce the monoalkynyl quinoline intermediate (Y). A further alkynylation of this intermediate provided the dialkynyl product 3. Though the nature of catalytic species generated in situ is not known when the reaction was performed in the absence of PPh3 affording the desired product in 15% yield (Entry 3, Table 1), Pd/C-mediated Sonogashira coupling of aryl iodides with terminal alkynes in the absence of ligand has been reported earlier.12 Nevertheless, a similar dialkynylation reaction was observed when 2,4-dichloroquinazoline was reacted with a terminal alkyne in the presence of (PPh3)2PdCl2, CuI and Et3N for 20 h.13 Notably, 2,4-dichloroquinoline did not participate in a dual dialkynylation reaction due to the lack of reactivity of C-4 chloro group.8,9 We overcame this issue in the present reaction by opting for a more reactive iodo group over chloro on the quinoline ring. Thus judicious selection of quinoline halide can help in affording a product of particular choice.
Conclusions
In conclusion, we have described a practical and one-step synthesis of 2,4-dialkynylquinoline from 2,4-diiodoquinoline and commercially available terminal alkynes under Pd/C-Cu catalysis in water. The reaction proceeds via dual C-C bond forming reaction without generating any side products. All the desired products were isolated in excellent yields. The reaction does not involve the use of expensive catalysts or solvents. Since the product nature can be altered conveniently by changing the halide moiety of the quinoline ring hence overall, the present process would be complimentary to the methods previously reported for alkynylation of quinolines. Due to the operational simplicity and easy availability of starting materials we believe that this process would find wide usage in the preparation of quinoline-based libraries of pharmaceutical interest.
Experimental
General methods
Unless stated otherwise, reactions were monitored by thin layer chromatography (TLC) on silica gel plates (60 F254), visualizing with ultraviolet light or iodine spray. Flash chromatography was performed on silica gel (60-120 mesh) using distilled petroleum ether and ethyl acetate. 1H and 13C NMR spectra were determined in CDCl3 solution using 400 and 50 MHz spectrometers, respectively. Proton chemical shifts (δ) are relative to tetramethylsilane (TMS, δ 0.0) as internal standard and expressed in parts per million. Spin multiplicities are given as s (singlet), δ (doublet), t (triplet), and m (multiplet) as well as b (broad). Coupling constants (J) are given in hertz. Infrared spectra were recorded on a FTIR spectrometer. Melting points were determined by using thermal analysis and differential scanning calorimetry (DSC) was generated with the help of DSC-60A detector. MS spectra were obtained on a mass spectrometer. Chromatographic (HPLC) purity was determined by using area normalization method and the condition specified in each case: column, mobile phase (range used), flow rate, detection wavelength, and retention times. Elemental analyses were performed using C,H,N elemental analyzer. All the reagents used are commercially available.
Preparation of 2,4-diiodoquinoline, 2 14
To a cold solution (5-10 ºC) of 2,4-dichloroquinoline (5.0 g, 25.3 mmol, 1.0 equiv.) in acetonitrile (50 mL) was added acetyl chloride (6.0 g, 76.4 mmol, 3.0 equiv.) and sodium iodide (30.0 g, 200.1 mmol, 8.0 equiv.). The mixture was stirred at 5-10 ºC for 30 min and slowly heated to reflux. The mixture was stirred at refluxing temperature for 6 h under nitrogen atmosphere and the progress of reaction was monitored by TLC. After completion of the reaction the mixture was cooled to room temperature and diluted with 10% Na2S2O3 to adjust the pH to 8.0-8.5. The solid precipitated out was filtered and was purified by column chromatography to give the desired product as a light brown solid (7.2 g, yield 75%); mp 89-90 ºC (lit14 90 ºC); 1H NMR (CDCl3, 400 MHz) δ 8.30 (s, 1H, Ar-H), 7.98-7.95 (m, 2H, Ar-H), 7.75-7.71 (m, 1H, Ar-H), 7.65-7.61 (m, 1H,Ar-H); m/z (ES Mass) 382 (M+1, 100%).
General procedure for the preparation of dialkynyl quinolines, 3
A mixture of 2,4-diiodoquinoline (2), 10% Pd/C (0.26 equiv.), CuI (0.05 equiv.), PPh3 (0.20 equiv.) and triethylamine (3.0 equiv.) in water (5.0 mL) was stirred at room temperature for 30 min. To this mixture was added terminal alkyne (3.0 equiv.) with stirring. The mixture was then stirred at 80-85 ºC for time mentioned in Table 1. After completion of the reaction (indicate by TLC) the reaction mass was cooled to room temperature, filtered through celite and extracted with ethyl acetate (2 × 30 mL). The organic layers were collected, washed with water (3 × 30 mL), dried over anhydrous Na2SO4, and concentrated. The crude residue obtained was purified by column chromatography on silica gel, using light petroleum (60-80 ºC)-ethyl acetate to afford the desired product 3.
2,4-di(Oct-1-ynyl)quinoline, 3a
Light brown liquid; Rf (20% ethyl acetate/n-hexane) 0.18; 1H NMR (CDCl3, 400 MHz) δ 8.19 (d, J 8.3 Hz, 1H, CH arom), 8.04 (d, J 8.2 Hz, 1H, CH arom), 7.71-7.67 (m, 1H, CH arom), 7.56-7.52 (m, 1H, CH arom), 7.50 (s, 1H), 2.57 (t, J 7.0 Hz, 2H, CH2), 2.48 (t, J 7.3 Hz, 2H, CH2), 1.74-1.28 (m, 4H, CH2), 1.25-1.12 (m, 12H, CH2), 0.99-0.82 (m, 6H, CH3); IR (neat) νmax/cm-1: 2926, 2227 (-C≡C-), 1581, 1463; m/z (ES Mass) 346 (M+1, 100%); 13CNMR (CDCl3, 50 MHz) 147.8 (C arom), 143.5 (C arom), 130.8 (C arom), 129.9 (2C, C arom), 128.3 (2C, C arom), 126.9 (C arom), 125.7 (C arom), 92.3 (2C, Csp), 80.7 (Csp), 77.6 (Csp), 37.0 (CH2), 36.6 (CH2), 34.3 (CH2), 32.7 (CH2), 31.9 (CH2), 30.3 (CH2), 29.3 (CH2), 28.4 (CH2), 27.9 (CH2), 22.6 (CH2), 19.5 (CH3), 14.3 (CH3); HRMS (ESI): Calc. for C25H32N (M+H)+ 346.2535; found 346.2542; Elemental analysis found: C, 86.85; H, 9.07; N, 4.14 C25H31N; Requires C, 86.90, H, 9.04, N, 4.05.
2,4-di(Hept-1-ynyl)quinoline, 3b
Light brown liquid; Rf (20% ethyl acetate/n-hexane) 0.26; 1H NMR (CDCl3, 400 MHz) δ 8.2 (d, J 8.1 Hz, 1H, CH arom), 8.03 (d, J 8.5 Hz, 1H, CH arom), 7.71-7.67 (m, 1H, CH arom), 7.56-7.52 (m, 1H, CH arom), 7.50 (s, 1H, CH arom), 2.57 (t, J 7.0 Hz, 2H, CH2), 2.48 (t, J 7.3 Hz, 2H, CH2), 1.75-1.65 (m, 4H, CH2), 1.50-1.42 (m, 8H, CH2), 0.97-0.86 (m, 6H, CH3); IR (neat) νmax/cm-1: 2929, 2227 (-C≡C-), 1581, 1357; m/z (ES Mass) 318 (M+1,100%); 13C NMR (CDCl3, 50 MHz), 147.8 (C arom), 143.4 (C arom), 130.7 (C arom), 129.8 (2C, C arom), 126.8 (3C, C arom), 125.5 (C arom), 92.1 (2C, Csp), 80.6 (Csp), 77.6 (Csp), 31.8 (2C, CH2), 29.6 (2C, CH2), 28.1 (CH2), 27.9 (CH2), 22.6 (2C, CH3), 19.6, 14.0; HRMS (ESI): Calc. for C23H28N (M+H)+ 318.2222; found 318.2231; Elemental analysis found: C, 87.11; H, 8.29; N, 4.24 C23H27N; Requires C, 87.02, H, 8.57, N, 4.41.
2,4-di(Hex-1-ynyl)quinoline, 3c
Light brown liquid; Rf (20% ethyl acetate/n-hexane) 0.22; 1H NMR (CDCl3, 400 MHz) δ 8.20 (d, J 8.1 Hz, 1H, CH arom), 8.05 (d, J 8.5 Hz, 1H, CH arom), 7.71-7.67 (m, 1H, CH arom), 7.57-7.52 (m, 1H, CH arom), 7.50 (s, 1H, CH arom), 2.58 (t, J 7.1 Hz, 2H, CH2), 2.49 (t, J 7.1 Hz, 2H, CH2), 1.69-1.48 (m, 8H, CH2), 0.99-0.89 (m, 6H, CH3); IR (neat) νmax/cm-1: 2927, 2227 (-C≡C-), 1581, 1357; m/z (ES Mass) 290 (M+1, 100%); 13C NMR (CDCl3, 50 MHz) 147.8 (C arom), 143.5 (C arom), 130.7 (C arom), 129.8 (C arom), 129.5 (C arom), 126.9 (3C, C arom), 125.6 (C arom), 92.1 (2C, Csp), 80.7 (Csp), 77.6 (Csp), 34.1 (CH2), 33.9 (CH2), 31.8 (CH2), 30.5 (CH2), 29.4 (CH2), 27.1 (CH2), 19.4 (CH3), 14.0 (CH3); HRMS (ESI): Calc. for C21H24N (M+H)+ 290.1909; found 290.1895; Elemental analysis found: C, 87.30; H, 8.00; N, 4.74 C21H23N; Requires C, 87.15, H, 8.01, N, 4.84.
2,4-bis(Phenylethynyl)quinoline, 3d
Pale yellow solid; mp 89-95 ºC; Rf (20% ethyl acetate/n-hexane) 0.20; 1H NMR (CDCl3, 400 MHz) δ 8.35 (d, J 8.5 Hz, 1H, CH arom), 8.16 (d, J 8.3 Hz, 1H, CH arom), 7.79-7.74 (m, 2H, CH arom), 7.69-7.61 (m, 5H, CH arom), 7.45-7.35 (m, 6H, CH arom); IR (neat) νmax/cm-1: 2926, 2214 (-C≡C-), 1579, 1369; m/z (ES Mass) 330 (M+1, 100%); 13C NMR (CDCl3, 50 MHz) 147.9 (C arom), 142.9 (C arom), 132.1 (4C, (C arom), 131.8 (2C, C arom), 130.3 (2C, C arom), 129.5 (3C, C arom), 128.4 (2C, C arom), 127.5 (2C, C arom), 126.6 (2C, C arom), 125.6 (C arom), 121.9 (C arom), 90.3 (2C, Csp), 88.8 (2C, Csp); HRMS (ESI): calc. for C25H16N (M+H)+ 330.1439; found 330.1459; Elemental analysis found: C, 91.30; H, 4.55; N, 4.21 C25H15N; Requires C, 91.16, H, 4.59, N, 4.25.
2,4-bis(p-Tolylethynyl)quinoline, 3e
Pale yellow solid; mp 150-153 ºC; Rf (20% ethyl acetate/n-hexane) 0.24 1H NMR (CDCl3, 400 MHz) δ 8.34 (d, J 8.07 Hz, 1H, CH arom) 8.14 (d, J 8.5 Hz, 1H, CH arom), 7.77 (s, 1H, CH arom), 7.75 (d, J 6.5 Hz, 1H, CH arom), 7.63-7.55 (m, 4H, CH arom), 7.25-7.18 (m, 5H, CH arom), 2.41 (s, 3H, CH3), 2.39 (s, 3H, CH3); IR (neat) νmax/cm-1: 2918, 2206 (-C≡C-), 1577, 1363; m/z (ES Mass) 358 (M+1, 100%); 13C NMR (CDCl3, 50 MHz) 148.13 (C arom), 143.27 (C arom), 139.4 (2C, C arom), 132.1 (2C, C arom), 131.8 (2C, C arom), 130.2 (C arom), 129.4 (4C, C arom), 127.39 (3C, C arom), 126.6 (2C, C arom), 125.7 (C arom), 118.9 (2C, C arom), 90.6 (2C, Csp), 88.5 (2C, Csp), 21.6 (2C, CH3); HRMS (ESI): calc. for C27H20N (M+H)+ 358.1596; found 358.1600; Elemental analysis found: C, 90.83; H, 5.40; N, 3.84 C27H19N; Requires C, 90.72, H, 5.36, N, 3.92.
2,4-bis(4-Phenylbut-1-ynyl)quinoline, 3f
Low melting solid; Rf (20% ethyl acetate/n-hexane) 0.25; 1H NMR (CDCl3, 400 MHz) δ 8.04 (d, J 8.2 Hz, 1H, CH arom), 7.9 (d, J 8.1 Hz, 1H, CH arom), 7.7-7.68 (m, 1H, CH arom), 7.50-7.48 (m, 1H, CH arom), 7.42 (s, 1H, CH arom), 7.37-7.23 (m, 10H, CH arom), 3.04-2.99 (m, 4H, CH2), 2.91-2.87 (m, 2H, CH2), 2.80-2.76 (m, 2H, CH2); IR (neat) νmax/cm-1: 2927, 2227 (-C≡C-), 1581, 1359; m/z (ES Mass) 386 (M+1, 100%); 13C NMR (CDCl3, 50 MHz) 147.7 (C arom), 143.1 (C arom), 140.2 (2C) (C arom), 130.0 (4C, C arom), 128.4 (3C, C arom), 127.0 (4C, C arom), 126.4 (4C, (C arom), 125.7 (2C, C arom), 91.2 (2C, Csp), 81.2 (2C, Csp), 34.6 (2C, CH2), 21.5 (2C, CH2); HRMS (ESI): Calc. for C29H24N (M+H)+ 386.1909; found 386.1907; Elemental analysis found: C, 90.19; H, 6.00; N, 3.94 C29H23N; Requires C, 90.35, H, 6.01, N, 3.63.
6,6'-(Quinoline-2,4-diyl)dihex-5-ynenitrile, 3g
Light brown oil; Rf (20% ethyl acetate/n-hexane) 0.19; 1H NMR (CDCl3, 400 MHz) δ 8.19 (d, J 7.1 Hz, 1H, CH arom), 8.05 (d, J 8.1 Hz, 1H, CH arom), 7.75-7.72 (m, 1H, CH arom), 7.61-7.58 (m, 1H, CH arom), 7.53 (s, 1H, CH arom), 2.82-2.78 (m, 2H, CH2), 2.71-2.64 (m, 2H, CH2), 2.62-2.59 (m, 4H, CH2), 2.14-2.0 (m, 4H, CH2); IR (neat) νmax/cm-1: 2927, 2247, 2229 (-C≡C-), 1581, 1359; m/z (ES Mass) 312 (M+1, 100%); 13C NMR (CDCl3, 50 MHz) 147.6 (C arom), 142.5 (C arom), 130.1 (C arom), 129.9 (2C, C arom), 128.5 (C arom), 127.4 (C arom), 126.3 (C arom), 125.3 (C arom), 118.6 (2C, C arom), 88.6 (2C, Csp), 81.9 (2C, Csp), 38.4 (CH2), 33.8 (CH2), 31.7 (CH2), 30.1 (CH2), 28.7 (CH2), 24.6 (CH2); HRMS (ESI): Calc. for C21H18N3 (M+H)+ 312.1501; found 312.1493; Elemental analysis found: C, 81.21; H, 5.49; N, 13.28 C21H17N3; Requires C, 81.00, H, 5.50, N, 13.49.
1,1'-(Quinoline-2,4-diylbis(ethyne-2,1-diyl)dicyclohexanol, 3h
Yellow solid, mp 154-163ºC, Rf (20% ethyl acetate/n-hexane); 0.19 1H NMR (CDCl3, 400 MHz) δ 8.19 (d, J 8.1 Hz, 1H, CH arom), 8.09 (d, J 8.1 Hz, 1H, CH arom), 7.74-7.70 (m, 1H, CH arom), 7.61-7.59 (m, 1H, CH arom), 7.58 (s, 1H, CH arom), 2.36-2.31 (bs, 2H, OH), 2.14-2.04 (m, 4H), 1.84-1.62 (m, 16H); IR (neat) νmax/cm-1: 3375 (C arom), 3062, 2929, 2222 (-C≡C-), 1583; m/z (ES Mass) 374 (M+1, 100%); 13C NMR (CDCl3, 50 MHz) 147.8 (C arom), 142.6 (C arom), 130.3 (C arom), 129.7 (2C, C arom), 127.6 (2C, C arom), 126.6 (C arom), 125.5 (C arom), 94.3 (2C, Csp), 83.7 (2C, Csp), 39.8 (C-OH), 39.6 (C-OH), 29.7 (3C, CH2), 25.1 (3C, CH2), 23.4 (3C, CH2), 23.2 (CH2); HRMS (ESI): Calc. for C25H28NO2 (M+H)+ 374.2120; found 374.2115; Elemental analysis found: C, 80.56; H, 7.30; N, 3.67 C25H27NO2; Requires C, 80.40, H, 7.29, N, 3.75.-
1,1'-(Quinoline-2,4-diyl)dipent-1-yn-3-ol, 3i
Light brown oil; Rf (30% ethyl acetate/n-hexane) 0.19; 1H NMR (CDCl3, 400 MHz) δ 8.16 (d, J 7.6 Hz, 1H, CH arom), 8.07 (d, J 8.6 Hz, 1H, CH arom), 7.72-7.68 (m, 1H, CH arom), 7.58-7.56 (m, 1H, CH arom), 7.50 (s, 1H, CH arom), 3.98-3.90 (m, 4H, CH2), 2.70-2.65 (m, 2H, CH2), 2.65-2.69 (m, 2H, CH2), 2.30-2.05 (bs, 2H, OH), 1.95-1.85 (m, 4H, CH2); IR (neat) νmax/cm-1: 3352, 3091, 2964, 2225 (-C≡C-), 1583; m/z (ES Mass) 294 (M+1, 100%); 13C NMR (CDCl3, 50 MHz) 147.5 (C arom), 142.3 (C arom), 130.6 (2C, C arom), 129.8 (C arom), 127.6 (2C, C arom), 126.6 (C arom), 125.5 (C arom), 91.9 (2C, Csp), 84.0 (2C, Csp), 64.4 (2C, CH2), 31.9 (CH2), 30.5 (CH2), 29.3 (CH2), 24.8 (CH2); HRMS (ESI): calc. for C19H20NO2 (M+H)+ 294.1494; found 294.1480; Elemental analysis found: C, 77.82; H, 6.51; N, 4.70 C19H19NO2; Requires C, 77.79, H, 6.53, N, 4.77.
5,5'-(Quinoline-2,4-diyl)dipent-4-yn-1-ol, 3j
Light brown liquid; Rf (30% ethyl acetate/n-hexane) 0.19; 1H NMR (CDCl3, 400 MHz) δ 8.17 (d, J 8.1 Hz, 1H, CH arom), 8.05 (d, J 8.2 Hz, 1H, CH arom), 7.7-7.67 (m, 1H, CH arom), 7.55-7.52 (m, 1H, CH arom), 7.47 (s, 1H, CH arom), 4.89-4.65 (m, 1H, CH(OH)), 4.65-4.60 (m, 1H, CH(OH)), 1.98-1.88 (m, 4H, CH2), 1.65-1.60 (bs, 2H, OH), 1.20-1.05 (m, 6H, CH3); IR (neat) νmax/cm-1: 3319 (broad), 2927, 2229 (-C≡C-), 1583, 1361; m/z (ES Mass) 294 (M+1, 100%); 13C NMR (CDCl3, 50 MHz) 147.5 (C arom), 143.0 (C arom), 130.8 (2C, C arom), 128.7 (C arom), 127.1 (C arom), 126.6 (2C, C arom), 125.6 (C arom), 91.8 (2C, Csp), 80.8 (2C, Csp), 61.0 (CH), 60.9 (CH), 31.8 (CH2), 30.8 (CH2), 28.8 (CH3), 24.7 (CH3); HRMS (ESI): Calc. for C19H20NO2 (M+H)+ 294.1494; found 294.1480; Elemental analysis found: C, 77.53; H, 6.52; N, 4.79 C19H19NO2; Requires C, 77.79, H, 6.53, N, 4.77.
Acknowledgments
The authors thank Dr. V. Dahanukar and Mr. A. Mukherjee for their encouragement and the analytical group for spectral data. Mr. E.A.R. thanks CPS-DRL, Hyderabad, India for allowing him to pursue this work as a part of his Ph.D. program.
References
1. Balasubramanian, M.; Keay, J. G. In Comprehensive Heterocyclic Chemistry II; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., eds.; Pergamon: Oxford, 1996; Vol. 5, pp. 245-300.
2. Claret, P. A.; Osborne, A. G. In The Chemistry of Heterocyclic Compounds; Quinolines; Jones, G., ed.; John Wiley and Sons: Chichester, UK, 1982; Vol. 32, Part 2, pp. 31, 32.
3. Daines, R. A.; Kingsbury, W. D .; Pendrak, I.; Mallamo, J. P.; U.S. Patent Application U.S. 6,100,267, 2000.
4. Martínez-Grueiro, M.; Giménez-Pardo, C.; Gómez-Barrio, A.; Franck, X.; Fournet, A.; Hocquemiller, R.; Figadère, B.; Casado-Escribano, N.; IL Farmaco 2005, 60, 219 and references cited therein.
5. Sonogashira, K.; Tohda, Y.; Hagihara, N.; Tetrahedron Lett. 1975, 4467.
6. Chinchilla, R.; Nájera, C.; Chem. Rev. 2007, 107, 874; Singh, R.; Sharma, M.; Mamgain, R.; Rawat, D . S.; J. Braz. Chem. Soc. 2008, 19, 357; Bakherad, M.; Keivanloo, A.; Bahramian, B.; Kamali, T. A.; J. Braz. Chem. Soc. 2009, 20, 907; Amaral, P. A.; Petrignet, J.; Gouault, N.; Agustini, T.; Lohézic-Ledévéhat, F.; Cariou, A.; Grée, R.; Eifler-Lima, V. L.; David, M.; J. Braz. Chem. Soc. 2009, 20, 1687.
7. Comins, D . L.; Nolan, J. M.; Bori, I. D .; Tetrahedron Lett. 2005, 46, 6697.
8. Reddy, E.A.; Islam, A.; Mukkanti, K.; Bandameedi, V.; Bhowmik, D . R.; Pal, M.; Beilstein; J. Org. Chem. 2009, 5, 32.
9. Reddy, E. A.; Barange, D . K.; Islam, A.; Mukkanti, K.; Pal, M.; Tetrahedron 2008, 64, 7143.
10. Chessa, G.; Canovese, L.; Visentin, F.; Santo, C.; Seragila, R.; Tetrahedron 2005, 61, 1755.
11. Pal, M.; Synlett 2009, 2896.
12. Mori, S.; Yanase, T.; Aoyagi, S.; Monguchi, Y.; Maegawa, T.; Sajiki, H.; Chem. Eur. J. 2008, 14, 6994.
13. Mangalagiu, I.; Benneche, T.; Undheim, K.; Acta Chem. Scand. 1996, 50, 914.
14. Bissember, A. C.; Banwell, M. G.; J. Org. Chem. 2009, 74, 4893; Chessa, G.; Canovese, L.; Visentin, F.; Santo, C.; Seraglia, R.; Tetrahedron 2005, 61, 1755.
Submitted: March 12, 2010
Published online: May 20, 2010
- 1. Balasubramanian, M.; Keay, J. G. In Comprehensive Heterocyclic Chemistry II; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., eds.; Pergamon: Oxford, 1996; Vol. 5, pp. 245-300.
- 2. Claret, P. A.; Osborne, A. G. In The Chemistry of Heterocyclic Compounds; Quinolines; Jones, G., ed.; John Wiley and Sons: Chichester, UK, 1982; Vol. 32, Part 2, pp. 31, 32.
- 3. Daines, R. A.; Kingsbury, W. D .; Pendrak, I.; Mallamo, J. P.; U.S. Patent Application U.S. 6,100,267, 2000
- 4. Martínez-Grueiro, M.; Giménez-Pardo, C.; Gómez-Barrio, A.; Franck, X.; Fournet, A.; Hocquemiller, R.; Figadère, B.; Casado-Escribano, N.; IL Farmaco 2005, 60, 219 and references cited therein.
- 5. Sonogashira, K.; Tohda, Y.; Hagihara, N.; Tetrahedron Lett 1975, 4467.
- 6. Chinchilla, R.; Nájera, C.; Chem. Rev 2007, 107, 874;
- Singh, R.; Sharma, M.; Mamgain, R.; Rawat, D . S.; J. Braz. Chem. Soc. 2008, 19, 357;
- Bakherad, M.; Keivanloo, A.; Bahramian, B.; Kamali, T. A.; J. Braz. Chem. Soc. 2009, 20, 907;
- Amaral, P. A.; Petrignet, J.; Gouault, N.; Agustini, T.; Lohézic-Ledévéhat, F.; Cariou, A.; Grée, R.; Eifler-Lima, V. L.; David, M.; J. Braz. Chem. Soc. 2009, 20, 1687.
- 7. Comins, D . L.; Nolan, J. M.; Bori, I. D .; Tetrahedron Lett. 2005, 46, 6697.
- 8. Reddy, E.A.; Islam, A.; Mukkanti, K.; Bandameedi, V.; Bhowmik, D . R.; Pal, M.; Beilstein; J. Org. Chem. 2009, 5, 32.
- 9. Reddy, E. A.; Barange, D . K.; Islam, A.; Mukkanti, K.; Pal, M.; Tetrahedron 2008, 64, 7143.
- 10. Chessa, G.; Canovese, L.; Visentin, F.; Santo, C.; Seragila, R.; Tetrahedron 2005, 61, 1755.
- 11. Pal, M.; Synlett 2009, 2896.
- 12. Mori, S.; Yanase, T.; Aoyagi, S.; Monguchi, Y.; Maegawa, T.; Sajiki, H.; Chem. Eur. J. 2008, 14, 6994.
- 13. Mangalagiu, I.; Benneche, T.; Undheim, K.; Acta Chem. Scand. 1996, 50, 914.
- 14. Bissember, A. C.; Banwell, M. G.; J. Org. Chem 2009, 74, 4893;
- Chessa, G.; Canovese, L.; Visentin, F.; Santo, C.; Seraglia, R.; Tetrahedron 2005, 61, 1755.
Publication Dates
-
Publication in this collection
10 Sept 2010 -
Date of issue
2010
History
-
Accepted
20 May 2010 -
Received
12 Mar 2010