Abstracts
Two new flavonoids, 3,4,10-trihydroxy-9-methoxypterocarpan and 2',4'-dihydroxy-4-methoxy-b-hydroxychacanonol, were isolated from Platymiscium floribundum, in addition to the homopterocarpin, 2',4',4-trihydroxychalcone, 7,3',5'-trihydroxyflavanone, 7,3'-dihydroxy-8,5'-dimethoxyisoflavanone, 8-hydroxy-5,6,7,-trimethoxycoumarin, 6-hydroxy-7,8-dimethoxycoumarin, 6,7,8-trimethoxycoumarin, 6,7-dimethoxycoumarin, 8-hydroxy-6,7-dimethoxycoumarin, 3β-acetoxy-28-hydroxyolean-12-ene, 1,2,3-trimethoxy-5-allyl-benzene, 3,4-dimethoxycinnamaldehyde, and β-sitosterol and stigmasterol as a mixture. Structural characterization of all compounds was established on the basis of spectroscopic methods, particularly 1D and 2D nuclear magnetic resonance (NMR) and comparison with data from literature.
flavonoids; coumarins; Platymiscium floribundum; Fabaceae
Dois novos flavonoides, 3,4,10-tri-hidróxi-9-metoxi-pterocarpano and 2',4'-di-hidróxi-4'-metoxi-β-hidroxichalcanonol, foram isolados a partir de Platymiscium floribundum, em adição a homopterocapina, 2',4',4-tri-hidroxichalcona, 7,3',5'-tri-hidroxiflavanona, 7,3'-di-hidróxi-8,5'-di-metoxiisoflavanona, 8-hidróxi-5,6,7-tri-metoxicumarina, 6-hidróxi-7,8-di-metoxicumarina, 6,7,8-tri-metoxicumarina, 6,7-di-metoxicumarina, 8-hidróxi-6,7-di-metoxicumarina, 3β-acetóxi-28-hidróxi-olean-12-eno, 1,2,3-tri-metoxi-5-alilbenzeno, 3,4-di-metoxicinamaldeído, e β-sitosterol e estigmasterol como mistura. A caracterização estrutural de todos os compostos foi estabelecida com base em métodos espectroscópicos, particularmente ressonância magnética nuclear (RMN) 1D e 2D e comparação com dados da literatura.
ARTICLE
New flavonoids and coumarins from Platymiscium floribundum Vogel
Pérsio A. VelosoI; Antonia T. A. PimentaI; Francisco M. de SousaI; Maria José C. FalcãoII; Nilce V. GramosaI; José Nunes da Silva JuniorI; Edilberto R. SilveiraI; Mary Anne S. LimaI,* * e-mail: mary@dqoi.ufc.br
IDepartamento de Química Orgânica e Inorgânica, Centro de Ciências, Universidade Federal do Ceará, CP 12.200, 60021-940 Fortaleza-CE, Brazil
IIDepartamento de Química, Universidade Estadual do Ceará, CP 12.200, 60021-940 Fortaleza-CE, Brazil
ABSTRACT
Two new flavonoids, 3,4,10-trihydroxy-9-methoxypterocarpan and 2',4'-dihydroxy-4-methoxy-b-hydroxychacanonol, were isolated from Platymiscium floribundum, in addition to the homopterocarpin, 2',4',4-trihydroxychalcone, 7,3',5'-trihydroxyflavanone, 7,3'-dihydroxy-8,5'-dimethoxyisoflavanone, 8-hydroxy-5,6,7,-trimethoxycoumarin, 6-hydroxy-7,8-dimethoxycoumarin, 6,7,8-trimethoxycoumarin, 6,7-dimethoxycoumarin, 8-hydroxy-6,7-dimethoxycoumarin, 3β-acetoxy-28-hydroxyolean-12-ene, 1,2,3-trimethoxy-5-allyl-benzene, 3,4-dimethoxycinnamaldehyde, and β-sitosterol and stigmasterol as a mixture. Structural characterization of all compounds was established on the basis of spectroscopic methods, particularly 1D and 2D nuclear magnetic resonance (NMR) and comparison with data from literature.
Keywords: flavonoids, coumarins, Platymiscium floribundum, Fabaceae
RESUMO
Dois novos flavonoides, 3,4,10-tri-hidróxi-9-metoxi-pterocarpano and 2',4'-di-hidróxi-4'-metoxi-β-hidroxichalcanonol, foram isolados a partir de Platymiscium floribundum, em adição a homopterocapina, 2',4',4-tri-hidroxichalcona, 7,3',5'-tri-hidroxiflavanona, 7,3'-di-hidróxi-8,5'-di-metoxiisoflavanona, 8-hidróxi-5,6,7-tri-metoxicumarina, 6-hidróxi-7,8-di-metoxicumarina, 6,7,8-tri-metoxicumarina, 6,7-di-metoxicumarina, 8-hidróxi-6,7-di-metoxicumarina, 3β-acetóxi-28-hidróxi-olean-12-eno, 1,2,3-tri-metoxi-5-alilbenzeno, 3,4-di-metoxicinamaldeído, e β-sitosterol e estigmasterol como mistura. A caracterização estrutural de todos os compostos foi estabelecida com base em métodos espectroscópicos, particularmente ressonância magnética nuclear (RMN) 1D e 2D e comparação com dados da literatura.
Introduction
Platymiscium floribundum Vog. is a medium sized tree which belongs to the Fabaceae-Papilionoideae family. In the Brazilian Northeast, this plant is popularly known as "sacambu" and "jacaranda-do-litoral" and has been used by the local population in folk medicine as an anti-inflammatory agent.1 In addition, the wood has showed high commercial value in carpentry.2
Previous chemical investigation of P. floribundum led to the isolation of isoflavonoids and pterocarpans from the heartwood.1 Since then, several activities such as cytotoxic,1 antimitotic3 and apoptosis in HL-60 human leukemia cells4 of the flavonoids have been evaluated.
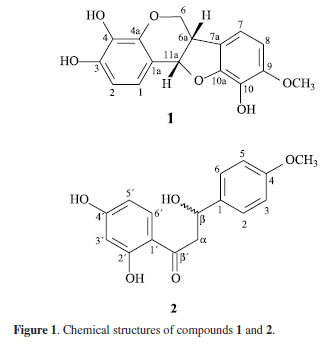
In continuation to the phytochemical investigation of heartwood, the study has been extended to trunk bark and roots of P. floribundum. This work reports on the isolation of two new flavonoids 3,4,10-trihydroxy-9-methoxypterocarpan (1) and 2',4'-dihydroxy-4-methoxy-β-hydroxychalcanonol (2) (Figure 1), in addition to the known homopterocarpin, 2',4',4-trihydroxychalcone, 7,3',5'-trihydroxyflavanone, 7,3'-dihydroxy-8,5'-dimethoxyisoflavanone, 8-hydroxy-5,6,7,-trimethoxycoumarin, 6-hydroxy-7,8-dimethoxycoumarin, 6,7,8-trimethoxycoumarin, 6,7-dimethoxycoumarin, 8-hydroxy-6,7-dimethoxycoumarin, 3β-acetoxy-28-hydroxyolean-12-ene, 1,2,3-trimethoxy-5-allyl-benzene, 3,4-dimethoxycinnamaldehyde, and β-sitosterol and stigmasterol as a mixture.
Results and Discussion
Compound 1 was obtained as a white solid with mp 108.2-110.0 ºC and  + 272º (CH3OH, c 0.02). The IR (infrared) spectrum showed absorption bands at 3379 and 1636 cm-1, characteristic of hydroxyl groups and aromatic skeleton, respectively. Its molecular formula was established by the negative mode HRESIMS (high-resolution electronspray ionization mass spectra) peak at m/z 301.0713 [M - H]- (calcd. for C16H14O6 - H, 301.0712).
+ 272º (CH3OH, c 0.02). The IR (infrared) spectrum showed absorption bands at 3379 and 1636 cm-1, characteristic of hydroxyl groups and aromatic skeleton, respectively. Its molecular formula was established by the negative mode HRESIMS (high-resolution electronspray ionization mass spectra) peak at m/z 301.0713 [M - H]- (calcd. for C16H14O6 - H, 301.0712).
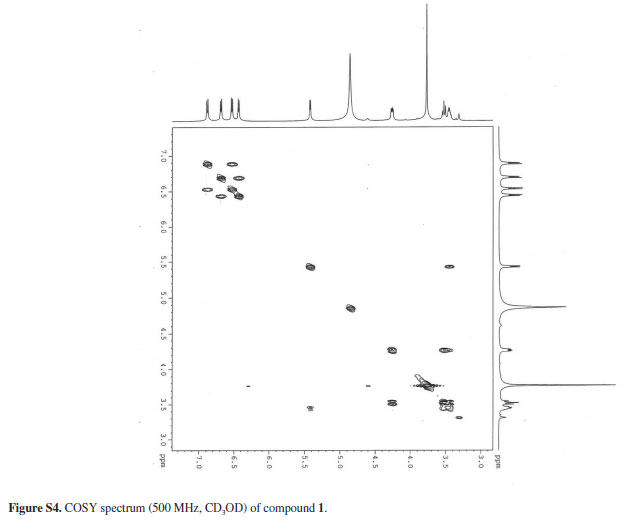
The 1H NMR spectrum suggested a pterocarpan structure due to the splitting pattern of the protons at δ 5.42 (d, J 6.6 Hz, H-11a), 4.26 (dd, J 4.7; 10.7 Hz, H-6α), 3.53 (t, J 10.7 Hz, H-6β) and δ 3.46 (m, H-6a), related to the protons of the heterocyclic ring B. This spectrum also allowed the identification of two pairs of ortho situated aromatic protons at δ 6.88 (d, J 8.4 Hz, H-1), 6.53 (d, J 8.4 Hz, H-2), 6.68 (d, J 8.1 Hz, H-7) and 6.43 (d, J 8.1 Hz, H-8), and gave a clear evidence of the 3,4,9,10-substitution pattern of the pterocarpan moiety. In addition, the presence of only one methoxyl group at δ 3.76 (s, OCH3) was consistent with a trihydroxylated pterocarpan skeleton.
All these data were supported by the 13C CPD (composite pulse decoupling) spectrum along with the 13C DEPT-135 (distortionless enhancement by polarization transfer) that revealed 16 carbon atoms corresponding to one methyl, one methylene, 6 methines and 8 non-hydrogenated carbons. However, the confirmation of the above suggestion for 1 was supported by the HSQC (heteronuclear single-quantum correlation spectroscopic) and HMBC (heteronuclear multiple-bond correlation spectroscopic) experiments, which allowed the unequivocal assignments of its 13C and 1H NMR data. The assignments of the relative positions of the methoxyl and the hydroxyl groups at C-9 and C-10, respectively, were defined in the HMBC spectrum that showed cross-peaks of the methyl protons at δ 3.79 (-OCH3) and the hydrogens at δ 6.68 (H-7) and 6.43 (H-8), with the carbon at d 150.53 (C-9), in addition to the correlation of the signal at d 6.43 (H-8) with the carbon at d 132.49 (C-10). Moreover, the correlation of hydrogen at δ 6.88 (H-1) with the carbons at δ 145.91 (C-4a), 80.70 (C-11a) and 147.19 (C-3), besides the correlations between the hydrogen at δ 6.53 (H-2) with the carbons at δ 147.19 (C-3), 113.91 (C-1a) definitively established that the other two hydroxyl groups were located at the C-3 and C-4 in the tetrassubstituted aromatic A-ring. Compound 1 is closely related to metilocarpan D, previously isolated from Metilotus alba5 except for the presence of one hydroxyl group at C-3 instead of a methoxyl group.
It is well known from the literature that, according to biogenetical regulations, the hydrogens (6a and 11a) at the B/C rings junction of all natural pterocarpans are always cis, either α,α or β,β, thus leading to only two enantiomeric forms. It is also known, through CD (circular dichroism) and/or ORD (optical rotatory dispersion) analyses, that (-) optical rotation can be associated with α,α positioning (6aR, 11aR), while the (+) optical rotation can be associated with the β,β positioning (6aS, 11aS) of both series.5 From the (+) optical rotation of compound 1, it could be assumed an (6aS, 11aS) absolute configuration for it. As expected, the CD spectrum of 1 should a similar profile of that from (+)-pterocarpin, and almost a mirror image of (-)-maackiain, what is in agreement with the suggested (6aS, 11aS) absolute stereochemistry for 1.5 Thus, it was possible to characterize 1 as the (6aS,11aS)-3,4,10-trihydroxy-9-methoxypterocarpan.
Compound 2, a white solid with mp 185.6-186.6 ºC and - 32º (CH3OH; c 0.02), showed molecular formula C16H16O5 that was established by the quasi-molecular ion at m/z 287.0979 [M - H]- (calcd. for C16H16O5 - H, 287.0919) in the HRESIMS spectrum. The absorption bands at 3418 and 1632 cm-1 in the IR spectrum were consistent with hydroxyl and carbonyl groups, respectively.
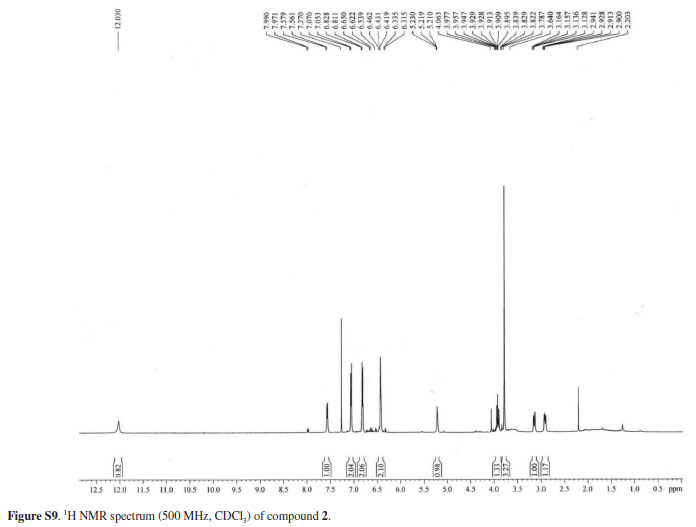
The 1H NMR spectrum showed hydrogens relative to one methoxyl group at δ 3.79 (s, -OCH3), a methylene at d 2.92 (dd, J 14.2, 6.3 Hz, H-ab) and 3.14 (dd, J 14.2, 3.8 Hz, H-aa), and a oxymethine hydrogen at δ 5.22 (dd, J 6.3, 3.8 Hz, H-β). The presence of a AA'BB' system in the aromatic region at δ 7.06 (d, J 8.5 Hz, H-2/6) and 6.82 (d, J 8.5 Hz, H-3/5) suggested the existence of a para-disubstituted benzene ring that was evidenced by the double intensity of the proton for two methines. The other aromatic ring was characterized as an ABX spin system, corresponding to a 1',2',4' substitution, by the signals at δ 7.57 (d, J 8.4 Hz, H-6'), 6.46 (d, J 8.4 Hz, H-5') and 6.43 (bs, H-3').
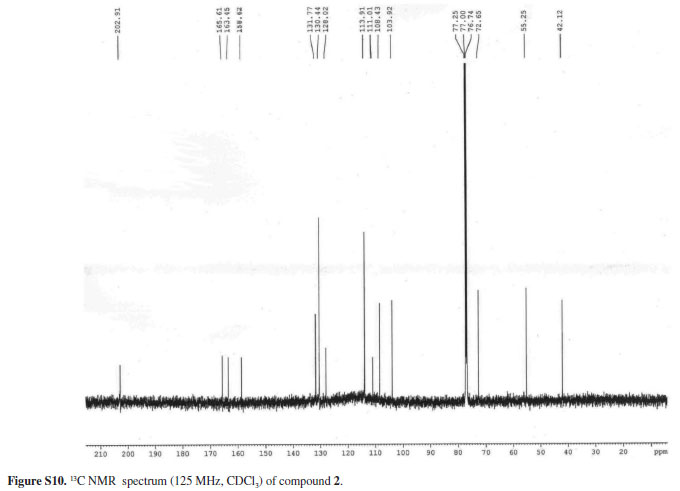
The 13C NMR spectrum showed 16 absorptions including one carbonyl, one methoxyl group, one methylene (δ 42.1), one oxymethine (δ 72.7) and twelve aromatic carbons (δ 103.9-165.3). On the basis of HMBC and HMQC spectral analysis, the structure of compound 2 was further determined, and all proton and carbon signals were fully assigned. In the HMBC spectrum, the hydrogens at δ 7.57 (H-6'), 3.14 (Hα-a) and 2.92 (Hα-b) correlated with the carbonyl carbon at d 202.9 (C-b'), the hydrogens at 7.57 (H-6'), and 6.43 (H-3') with the carbon at δ 165.6 (C-2'), and the hydrogens at δ 6.46 (H 3'/5') and 7.57 (H-6') with the carbon at δ 163.5 (C-4'). These findings indicated that the hydroxyl groups were located at C-2' and C-4' on the A ring in the chalcanonol skeleton. On the other hand, the long-range correlation observed between the aromatic hydrogens at δ 6.82 (H-3/5), 7.02 (H-2/6) and the methoxyl at δ 3.79 with the carbon at δ 158.6 (C-4) allowed the attachment of the methoxyl group to the C-4 carbon of the para-substituted aromatic ring. Thus, the structure of compound 2 was established as the new 2',4'-dihydroxy-4-methoxy-β-hydroxychalcanonol.
Experimental
General procedures
IR spectra were recorded on an ABB BOMEN FT-IR 1000 spectrometer, using KBr pellets. The NMR spectra were performed on a Bruker Avance DRX 500 spectrometer, equipped with an inverse detection probehead and z-gradient accessory working at 499.9 (1H) and at 124.97 MHz (13C), respectively. All pulse sequences are standard in the Bruker XWIN-NMR software, and all experiments were conducted at the room temperature. The samples, dissolved either in CDCl3, CD3OD or C5D5N (0.6 ml), were transferred to 5 mm tubes. High resolution mass spectra were recorded on an UltrOTOF-Q (quadrupole orthogonal time-of-flight) mass spectrometer (LC-IP-TOF model 225-07100-34, Shimadzu) by negative ionization mode of the ESI source. HPLC (high performance liquid chromatographic) analyses were performed on a Shimadzu chromatograph (Japan) equipped with a ternary pump (Shimadzu LC-20AT) and PDA (photodiode array) detector (Shimadzu SPD-M20A), using Phenomenex columns (analytical: 250 x 4.6 mm, 5 µm; semi-preparative: 250 x 10 mm, 10 µm). HPLC grade n-hexane, CHCl3 and EtOAc were purchased from Tedia Co (Brazil, São Paulo) and the HPLC grade water was obtained by a Milli-Q purification system (Millipore, Bedford, USA). Column chromatography was performed either over silica gel 60 (VETEC, 70-230 and 40-63 mesh) or Sephadex LH-20 (Amersham Pharmacia Biotech, Uppsala, Sweden). TLC (thin layer chromatograph) was performed on precoated silica gel aluminum sheets (Merck) and the compounds were visualized by UV detection and by spraying with vanillin/perchloric acid/EtOH solution, followed by 5 min heating.
Plant material
An entire P. floribundum specimen was collected in Acarape County, Ceará State, Brazil. A voucher specimen (No. 31052) was identified by Dr. Edson Paula Nunes and deposited at the Herbário Prisco Bezerra (EAC), Departamento de Biologia, Universidade Federal do Ceará, Fortaleza-CE, Brazil.
Extraction and isolation
The air dried roots (6.9 kg) of P. floribundum were pulverized and extracted with EtOH at room temperature. The solvent was removed under reduced pressure yielding a reddish resinous extract (1.8 kg). Part of the ethanol extract (350.0 g) was coarsely fractionated on a silica gel column by elution with n-hexane, CH2Cl2, EtOAc and MeOH. Chromatography of an aliquot of the CH2Cl2 fraction (20.0 g) on silica gel using n-hexane/EtOAc mixtures with increasing polarity yielded thirty three resulting fractions. The sub-fraction F-5 (123.0 mg) was purified by HPLC using n-hexane/EtOAc (68:32) (flow rate 4.72 ml min-1) to yield 8-hydroxy-5,6,7,-trimethoxycoumarin6 (45.6 mg), 6-hydroxy-7,8-dimethoxycoumarin7 (16.8 mg) and 2',4'-dihydroxy-4-methoxy-β-hydroxychalcanonol 2 (12.1 mg). The sub-fraction F-7 was recrystallized with methanol to yield the 6,7,8-trimethoxycoumarin8 (23.5 mg). The sub-fraction F-8 yielded homopterocarpin9 (15.5 mg) as a colorless solid. Flash chromatography of the sub-fraction F-9 (1.8 g) using n-hexane/CH2Cl2 (8:2) as solvent yielded 6,7-dimethoxycoumarin10 (96.5 mg). Recrystallization of subfraction F-13 with CH2Cl2 yielded 8-hydroxy-6,7-dimethoxycoumarin (190.3 mg).11
The air dried trunk bark (4.27 kg) was pulverized and extracted with EtOH at room temperature. The solvent was removed under reduced pressure to give a dark viscous extract (134.3 g). Liquid-liquid partition of the EtOH extract (134.3 g) using n-hexane, CH2Cl2 and EtOAc as solvents yielded four fractions. The CH2Cl2 fraction (21.3 g) was coarsely fractionated on a silica gel column by elution with n-hexane, CHCl3, EtOAc and MeOH, to yield twenty three fractions that were pooled together in four fractions after TLC analysis. The fraction F-1 (148.5 mg) was further purified by HPLC using a mixture of CHCl3/EtOAc (60/40; v/v) (flow rate 3.00 ml min-1) to afford 3,4,10-trihydroxy-9-methoxy-pterocarpan 1 (13.0 mg). Flash chromatography of the EtOAc fraction (11.4 g) using CHCl3/EtOAc mixtures with increasing polarity afforded five resulting fractions after TLC analysis. The subfraction F-3 (46.1 mg) was purified by HPLC using a mixture of CHCl3/EtOAc (60/40 v/v) (flow rate 3.0 ml min-1), to yield 7,3',5'-trihydroxyflavanone12 (9.8 mg) and 7,3'-dihydroxy-8,5'-dimethoxyisoflavanone (4.1 mg).13
Air-dried trunk heartwood (0.8 kg) was pulverized and extracted with n-hexane (3 x 3.0 L) at room temperature. The solvent was removed under reduced pressure yielding a viscous brown oil (7.0 g). The marc obtained after n-hexane extraction was extracted with CHCl3 (9.0 L) to give a reddish resinous extract (76.0 g). Finally, the residue was extracted with EtOH (3 x 3.0 L) to yield a viscous brown extract (35.0 g).
Part of the CHCl3 extract (60.0 g) was adsorbed onto silica gel (180.0 g) and coarsely fractionated over a silica gel column by elution with n-hexane, CHCl3, EtOAc and MeOH to give eight fractions. Fraction F-2 (6.9 g) was rechromatographed over a of silica gel column, and further eluted with an isocratic mixture of n-hexane/EtOAc (9:1) to give sixty five fractions, which were subsequently combined on the basis of TLC analysis to yield eleven fractions. Fraction F-2 gave the pure 6,7-dimethoxycoumarin (0.7 g).10 Fraction F-3 (400.0 mg) was rechromatographed on Sephadex LH-20 by elution with MeOH to give the 2',4',4-trihydroxychalcone14 (3.0 mg). Fraction F-4 (22.4 g) was rechromatographed over a column of silica gel, using n-hexane, CHCl3 and EtOAc mixtures with increasing polarity to yield five fractions. Flash chromatography of the sub-fraction F-4 (2) (120.0 mg) using CHCl3/EtOAc mixtures with increasing polarity afforded 3β-acetoxy-28-hydroxyolean-12-ene (10.0 mg).15 The subfraction F-4 (3) (370.0 mg) was further purified by flash chromatography after elution with n-hexane, CHCl3, EtOAc and MeOH with increasing polarity yielding a mixture of β-sitosterol and stigmasterol (22.0 mg).16 Flash chromatography of the sub-fraction F-4 (4) (170. 0 mg) by elution with CH2Cl2 yielded 7-methoxy-6-hydroxy-coumarin17 (15.0 mg) and 7-hydroxy-6-methoxy-coumarin (12.0 mg).17 The sub-fraction F-4 (5) (4.0 g) was further purified by flash chromatography after elution with mixtures of n-hexane and CHCl3 and EtOAc with increasing polarity to afford 6,7,8-trimethoxycoumarin (12.0 mg), 1,2,3-trimethoxy-5-allyl-benzene (10.0 mg) and 3,4-dimethoxycinnamaldehyde (10.0 mg).18,19
3,4,10-Trihydroxy-9-methoxypterocarpan (1)
White solid, mp 108.2-110.0 ºC; + 272º (c 0.002, MeOH); IR (KBr) νmax / cm-1 3379 (O-H), 1481, 1364, 1250, 1082; 1H NMR (500 MHz, CD3OD) 6.88 (d, J 8.4 Hz, H-1), 6.53 (d, J 8.4 Hz, H-2), 3.53 (t, J 10.6 Hz, H-6β), 4.26 (dd, J 4.7, 10.6 Hz, H-6α), 3.46 (m, H-6a), 3.76 (s, OCH3), 6.68 (d, J 8.1 Hz, H-7), 6.43 (d, J 8.1 Hz, H-8), 3.76 (m, H-OCH3); 13C NMR (125 MHz, CD3OD) 122.50 (C-1), 110.51 (C-2), 147.19 (C-3) 134.35 (C-4), 145.91 (C-4a), 67.92 (C-6), 41. 65 (C-6a), 122.90 (C-7a), 115.80 (C-7), 106.04 (C-8), 150.53 (C-9), 132.49 (C-10), 148.41 (C-10a), 80.70 (C-11a), 113.91 (C-1a), 57.1 (-OCH3); HRESIMS (negative ion mode) molecular ion peak at m/z 301.0752 [M - H]+ (calcd. for C16H13O6, 301.0712).
2',4'-Dihydroxy-4-methoxy-β-hydroxychalcanonol (2)
White solid, mp 185.6-186.6 ºC; - 32º (c 0.002, MeOH); IR (KBr) νmax / cm-1 3418 (O-H), 1632 (C=O), 1444, 1325, 1248,1178; 1H NMR (500 MHz, CDCl3) 7.06 (d, J 8.6 Hz, H-2), 6.82 (d, J 8.5 Hz, H-3), 6.82 (d, J 8.6 Hz, H-5), 7.06 (d, J 8.5 Hz, H-6), 3.79 (s, OCH3), 6.46 (s, H-3'), 6,46 (s, H-5'), 7.57 (d, J 9.4 Hz, H-6'), 5.22 (br t, J 5.6 Hz, H-β), 2,92 (dd, J 14.2, 6.3, Hz, H-αb), 3.14 (dd, J 14.2, 3.8 Hz, H-αa); 13C NMR (125 MHz, CDCl3) 128.00 (C-1), 130.44 (C-2), 113.9 (C-3) 158.62 (C-4), 113.91 (C-5), 130.4 (C-6), 111.01 (C-1'), 165.61 (C-2'), 103.92 (C-3'), 163.45 (C-4'), 108.43 (C-5'), 131.8 (C-6'), 72.65 (C-β), 42.1 (C-α), 202.9 (C-β'), 55.3 (-OCH3); HRESIMS (negative ion mode) molecular ion peak at m/z 287.0979 [M - H]+ (calcd. for C16H15O5, 287.0919).
Supplementary Information
Supplementary data are available free of charge at http://jbcs.sbq.org.br as PDF file.
Acknowledgments
The authors are grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamente de Pessoal de Nível Superior (CAPES), Programa de Apoio a Núcleos de Excelência (PRONEX) and Fundação Cearense de Apoio ao Desenvolvimento Científico e Tecnológico (FUNCAP) for the fellowships and financial support and to Dr. Darin Cesarin Sobrinho of the Universidade Federal Rural do Rio de Janeiro for the CD data.
References
1. Falcão, M. J.C.; Pouliquem, Y. B. M.; Lima, M. A. S.; Gramosa, N. V.; Costa-Lotufo, L. V.; Militão, G. C. G.; Pessoa, C.; Moraes, M. O.; Silveira, E. R.; J. Nat. Prod. 2005, 68, 423.
2. Lorenzi, H.; Árvores Brasileiras, vol. 1; Plantarum: São Paulo, 1992.
3. Militão, G. C. G.; Jimenez, P. C.; Wilke, D. V.; Pessoa, C.; Falcão, M. J. C.; Lima, M. A. S.; Silveira, E. R.; Moraes, M. O.; Costa-Lotufo, L. V.; Planta Med. 2005, 71, 683.
4. Militão, G. C. G.; Dantas, I. N. F.; Pessoa, C.; Falcão, M. J. C.; Silveira, E. R.; Lima, M. A. S.; Curi, R.; Lima, T.; Moraes, M. O.; Costa-Lotufo, L. V.; Life Sci. 2006, 78, 2409.
5. Cook, J. T.; Ollis, W. D.; Sutherland, I. O.; Gottlieb, O. T.; Phytochemistry 1978, 17, 1419. Sicherer, C. A. X. G. F.; Sicherer-Roetman, A.; Phytochemistry 1980, 19, 485. Miyase, T.; Ohtsubo, A.; Ueno, A.; Noro, T.; Kuroyanagi, M.; Fukushima, S.; Chem. Pharm. Bull. 1982, 30, 1986; Antus, S.; Kurtan, T.; Juhasz, L.; Kiss, L.; Hollosi, M.; Majer, S.; Chirality 2001, 13, 493.
6. Yun, B. S.; Lee, I. K.; Ryoo, I. J.; Yoo, I. D.; J. Nat. Prod. 2001, 64, 1238.
7. Gu, J. Q.; Graf, T. N.; Lee, D.; Chai, H. B.; Mi, Q.; Kardono, L. B. S.; Setyowati, F. M.; Ismail, R.; Riswan, S.; Farnsworth, N. R.; Cordell, G. A.; Pezzuto, J. M.; Swanson, S. M.; Kroll, D. J.; Falkinham, J. O.; Wall, M. E.; Wani, M. C.; Kinghorn, A. D.; Oberlies, N. H; J. Nat. Prod. 2004, 67, 1156.
8. Goh, S. H.; Chung, V. C.; Sha, C. K.; Mak, T. C. W.; Phytochemistry 1990, 29, 1704.
9. Pelter, A.; Ward, R. S.; Gray, T. I.; J. Chem. Soc., Perkin Trans.1 1976, 23, 2475.
10. Lee, S.; Kim, B. K.; Cho, S. H.; Shin, K. H.; Arch. Pharmacol. Res. 2002, 25, 280.
11. Aiyelaagbe, O. O.; Gloer, J. B.; Rec. Nat. Prod. 2008, 24, 100.
12. Qiong, Z.; Xue-Ting, L.; Jing-Yu, L.; Zhi-Da, M.; Chin. J. Nat. Med. 2008, 6, 168.
13. Shrestha, S. P.; Narukawa, Y.; Takeda T.; Chem. Pharm. Bull. 2007, 55, 926.
14. Markham, K. R.; Tetrahedron 1976, 32, 2607.
15. Ulubelen, A.; Topcu, G.; Fitoterapia 1987, 58, 205.
16. Aragão, P. C. A.; Toledo, J. B. T.; Morais, A. A.; Braz-Filho, R.; Quím. Nova 1990, 13, 254.
17. Zolek, T.; Paradowska, K.; Wawer, I.; Solid State Nucl. Magn. Reson. 2003, 23, 77.
18. Vasconcelos, J. M. J; Silva, A. M. S.; Cavaleiro, J. A. S.; Phytochemistry 1998, 49, 1421.
19. Burgi, C.V.; Gui, L.; Helv. Chim. Acta 1993, 76, 1901.
Submitted: October 18, 2011
Published online: June 12, 2012
Supplementary Information

Figure S1 - Click to enlarge

Figure S2 - Click to enlarge

Figure S3 - Click to enlarge

Figure S4 - Click to enlarge

Figure S5 - Click to enlarge

Figure S6 - Click to enlarge

Figure S7 - Click to enlarge

Figure S8 - Click to enlarge

Figure S9 - Click to enlarge

Figure S10 - Click to enlarge

Figure S11 - Click to enlarge

Figure S12 - Click to enlarge

Figure S13 - Click to enlarge

Figure S14 - Click to enlarge

Figure S15 - Click to enlarge
- 1. Falcão, M. J.C.; Pouliquem, Y. B. M.; Lima, M. A. S.; Gramosa, N. V.; Costa-Lotufo, L. V.; Militão, G. C. G.; Pessoa, C.; Moraes, M. O.; Silveira, E. R.; J. Nat. Prod. 2005, 68, 423.
- 2. Lorenzi, H.; Árvores Brasileiras, vol. 1; Plantarum: São Paulo, 1992.
- 3. Militão, G. C. G.; Jimenez, P. C.; Wilke, D. V.; Pessoa, C.; Falcão, M. J. C.; Lima, M. A. S.; Silveira, E. R.; Moraes, M. O.; Costa-Lotufo, L. V.; Planta Med. 2005, 71, 683.
- 4. Militão, G. C. G.; Dantas, I. N. F.; Pessoa, C.; Falcão, M. J. C.; Silveira, E. R.; Lima, M. A. S.; Curi, R.; Lima, T.; Moraes, M. O.; Costa-Lotufo, L. V.; Life Sci. 2006, 78, 2409.
- 5. Cook, J. T.; Ollis, W. D.; Sutherland, I. O.; Gottlieb, O. T.; Phytochemistry 1978, 17, 1419.
- Sicherer, C. A. X. G. F.; Sicherer-Roetman, A.; Phytochemistry 1980, 19, 485.
- Miyase, T.; Ohtsubo, A.; Ueno, A.; Noro, T.; Kuroyanagi, M.; Fukushima, S.; Chem. Pharm. Bull. 1982, 30, 1986;
- Antus, S.; Kurtan, T.; Juhasz, L.; Kiss, L.; Hollosi, M.; Majer, S.; Chirality 2001, 13, 493.
- 6. Yun, B. S.; Lee, I. K.; Ryoo, I. J.; Yoo, I. D.; J. Nat. Prod. 2001, 64, 1238.
- 7. Gu, J. Q.; Graf, T. N.; Lee, D.; Chai, H. B.; Mi, Q.; Kardono, L. B. S.; Setyowati, F. M.; Ismail, R.; Riswan, S.; Farnsworth, N. R.; Cordell, G. A.; Pezzuto, J. M.; Swanson, S. M.; Kroll, D. J.; Falkinham, J. O.; Wall, M. E.; Wani, M. C.; Kinghorn, A. D.; Oberlies, N. H; J. Nat. Prod. 2004, 67, 1156.
- 8. Goh, S. H.; Chung, V. C.; Sha, C. K.; Mak, T. C. W.; Phytochemistry 1990, 29, 1704.
- 9. Pelter, A.; Ward, R. S.; Gray, T. I.; J. Chem. Soc., Perkin Trans.1 1976, 23, 2475.
- 10. Lee, S.; Kim, B. K.; Cho, S. H.; Shin, K. H.; Arch. Pharmacol. Res. 2002, 25, 280.
- 11. Aiyelaagbe, O. O.; Gloer, J. B.; Rec. Nat. Prod 2008, 24, 100.
- 12. Qiong, Z.; Xue-Ting, L.; Jing-Yu, L.; Zhi-Da, M.; Chin. J. Nat. Med. 2008, 6, 168.
- 13. Shrestha, S. P.; Narukawa, Y.; Takeda T.; Chem. Pharm. Bull 2007, 55, 926.
- 14. Markham, K. R.; Tetrahedron 1976, 32, 2607.
- 15. Ulubelen, A.; Topcu, G.; Fitoterapia 1987, 58, 205.
- 16. Aragão, P. C. A.; Toledo, J. B. T.; Morais, A. A.; Braz-Filho, R.; Quím. Nova 1990, 13, 254.
- 17. Zolek, T.; Paradowska, K.; Wawer, I.; Solid State Nucl. Magn. Reson 2003, 23, 77.
- 18. Vasconcelos, J. M. J; Silva, A. M. S.; Cavaleiro, J. A. S.; Phytochemistry 1998, 49, 1421.
- 19. Burgi, C.V.; Gui, L.; Helv. Chim. Acta 1993, 76, 1901.
Publication Dates
-
Publication in this collection
03 Aug 2012 -
Date of issue
July 2012
History
-
Received
18 Oct 2011 -
Accepted
12 June 2012