Abstracts
Cyclic four-membered ring peroxides are important high-energy intermediates in a variety of chemi and bioluminescence transformations. Specifically, α-peroxylactones (1,2-dioxetanones) have been considered as model systems for efficient firefly bioluminescence. However, the preparation of such highly unstable compounds is extremely difficult and, therefore, only few research groups have been able to study the properties of these substances. In this study, the synthesis, purification and characterization of three 1,2-dioxetanones are reported and a detailed procedure for the known synthesis of diphenoyl peroxide, another important model compound for the chemical generation of electronically excited states, is provided. For most of these peroxides, the complete spectroscopic characterization is reported here for the first time.
organic peroxides; diphenoyl peroxide; 1,2-dioxetanones; α-peroxylactones; chemiluminescence
Peróxidos cíclicos de quatro membros são intermediários de alta energia importantes em diversas transformações quimi e bioluminescentes. Especificamente, α-peroxilactonas (1,2-dioxetanonas) têm sido consideradas sistemas modelo para a eficiente bioluminescência do vaga-lume. Contudo, a preparação deste tipo de compostos altamente instáveis é extremamente difícil e, por isso, apenas alguns poucos grupos de pesquisa puderam estudar as propriedades dessas substâncias. Neste trabalho, a síntese, purificação e caracterização de três 1,2-dioxetanonas são relatadas e é apresentado um procedimento detalhado para a preparação do peróxido de difenoíla, outro importante composto-modelo para a geração química de estados eletronicamente excitados. Para a maioria destes peróxidos, a caracterização espectroscópica completa é relatada pela primeira vez.
ARTICLE
Synthesis of unstable cyclic peroxides for chemiluminescence studies
Fernando H. Bartoloni## Current address: Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, 09210-170 Santo André-SP, Brazil;Marcelo A. de Oliveira; Felipe A. Augusto; Luiz Francisco M. L. Ciscato## Current address: Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, 09210-170 Santo André-SP, Brazil;Erick L. Bastos; Wilhelm J. Baader** e-mail: wjbaader@iq.usp.br
Departamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, 05508-000 São Paulo-SP, Brazil
ABSTRACT
Cyclic four-membered ring peroxides are important high-energy intermediates in a variety of chemi and bioluminescence transformations. Specifically, α-peroxylactones (1,2-dioxetanones) have been considered as model systems for efficient firefly bioluminescence. However, the preparation of such highly unstable compounds is extremely difficult and, therefore, only few research groups have been able to study the properties of these substances. In this study, the synthesis, purification and characterization of three 1,2-dioxetanones are reported and a detailed procedure for the known synthesis of diphenoyl peroxide, another important model compound for the chemical generation of electronically excited states, is provided. For most of these peroxides, the complete spectroscopic characterization is reported here for the first time.
Keywords: organic peroxides, diphenoyl peroxide, 1,2-dioxetanones, α-peroxylactones, chemiluminescence
RESUMO
Peróxidos cíclicos de quatro membros são intermediários de alta energia importantes em diversas transformações quimi e bioluminescentes. Especificamente, α-peroxilactonas (1,2-dioxetanonas) têm sido consideradas sistemas modelo para a eficiente bioluminescência do vaga-lume. Contudo, a preparação deste tipo de compostos altamente instáveis é extremamente difícil e, por isso, apenas alguns poucos grupos de pesquisa puderam estudar as propriedades dessas substâncias. Neste trabalho, a síntese, purificação e caracterização de três 1,2-dioxetanonas são relatadas e é apresentado um procedimento detalhado para a preparação do peróxido de difenoíla, outro importante composto-modelo para a geração química de estados eletronicamente excitados. Para a maioria destes peróxidos, a caracterização espectroscópica completa é relatada pela primeira vez.
Introduction
The light emission resulting from a chemical transformation is called chemiluminescence.1 Most chemiluminescent reactions2-5 have four-membered cyclic organic peroxides as high-energy intermediates. These compounds are fundamental for the chemical generation of electronic excited states1 and are assumed to take part also in bioluminescence reactions, such as the firefly luciferin/luciferase system.6,7
Many theoretical investigations have been performed, including recent studies, to contribute to the mechanistic elucidation of chemiluminescent and bioluminescent transformations.8-17 Furthermore, some of these unstable cyclic peroxides with high energy content have been prepared, allowing the experimental mechanistic investigation of chemiluminescent reactions8,18-20 as well as the development of several applications, including the uphill energy conversion.21 In 1969, Kopecky and Mumford22 synthesized 3,3,4-trimethyl-1,2-dioxetane (Scheme 1, 1: R1 = R2 = R3 = CH3, R4 = H), the first 1,2-dioxetane (1) derivative, a compound formerly assumed to be too unstable to be isolated. Three years later, Adam and Liu23 reported the synthesis of 3-tert-butyl-1,2-dioxetanone (Scheme 1, 2: R1 = tert-butyl, R2 = H), the first 1,2-dioxetanone (2) derivative, using the corresponding α-hydroperoxy carboxylic acid as precursor. These cyclic peroxides decompose thermally and can generate one of the two carbonyl fragments in its electronic excited state.1 However, the chemiluminescence emission efficiency of these unimolecular processes is low (ΦCL < 0.01%),1,24,25 because triplet-excited carbonyl compounds are formed preferentially (i.e., ΦS < 104 E mol1vs. ΦT up to 0.3 E mol1).26
Several cyclic peroxide derivatives have been prepared since the pioneering works of Kopecky and Mumford22 and Adam and Liu23, and it was found that fluorescent oxidizable compounds1 were able to catalyze the decomposition of some of them, e.g., diphenoyl peroxide (3),24,27,28 3,3-dimethyl-1,2-dioxetanone (4)29-35 (Scheme 2). These compounds have been used as simple chemical models in order to rationalize the efficient excited state generation in firefly bioluminescence (ΦBL ca. 0.4 E mol1).36,37 However, Catalani and Wilson38 found that compound 3 is very inefficient in generating electronically excited states upon catalyzed decomposition, an observation confirmed more recently by our research group.39 Additionally, our group found that compound 4, a much better model for the α-peroxylactone derivative formed in the bioluminescent transformation of firefly luciferin, is also highly inefficient for excited state formation upon catalyzed decomposition.39 Contrarily, there are other highly efficient chemiluminescent reactions involving cyclic peroxides, like the intramolecular decomposition of electron-rich 1,2-dioxetanes and the peroxyoxalate reaction, where a cyclic four-membered ring peroxide is believed to occur as an intermediate.1,40-53
In contrast to the convenient preparation of sterically-hindered 1,2-dioxetanes,54-56 the difficult synthesis and purification of 1,2-dioxetanones and related cyclic peroxides still limits the investigation of the relationship between their structure and the chemiexcitation efficiency.1,26 Only the research groups of W. Adam, G. B. Schuster and N. J. Turro have accomplished the synthesis of 1,2-dioxetanone derivatives; however, no other research group has ever reported the synthesis of any of these derivatives, indicating the extreme difficulties in working with this class of compounds.1
In this work, the synthesis, purification and characterization of diphenoyl peroxide (3), 3,3-dimethyl-1,2-dioxetanone (4), spiro-adamantyl-1,2-dioxetanone (5) and spiro-cyclopentyl-1,2-dioxetanone (6) are reported (Scheme 2). Literature data related to the synthesis and characterization for the known compounds 3-5 are sparse and lack experimental details, which are provided here (including details on the preparation and characterization of the α-hydroperoxyacid precursors 7-9). Furthermore, the complete NMR spectroscopic characterization of compounds 3 and 4, as well as the synthesis and characterization of the novel α-peroxylactone derivative 6 are described here for the first time. These synthesized compounds have been used for chemiluminescence emission quantum yield determinations and detailed mechanistic studies.39
Experimental
Materials and methods
Chemicals
Pentane (Synth), hexane (Synth) and CH2Cl2 (Synth) were stirred overnight over EDTA (ethylenediaminetetraacetate), filtered and distilled, thereafter distilled again from metallic sodium (alkanes) or P2O5 (CH2Cl2). Dimethylsulfoxide (DMSO, Synth) was heated to ca. 100 ºC, distilled under reduced pressure from CaH2 (Aldrich) and stored over 4 Å molecular sieves, under argon. MeOH (Synth) was refluxed and distilled (1 L) from magnesium methoxide (prepared from 50 mL of MeOH, 5 g of magnesium and 0.5 g of iodine). Et2O (Synth or Vetec) was refluxed and distilled from H2SO4 conc. (Merck, 100 mL per 1 L Et2O) and then refluxed and distilled from sodium/benzophenone (Acros). THF (Sigma-Aldrich) was refluxed over sodium and distilled from sodium/benzophenone. EtOAc (Sigma-Aldrich) was kept over CaCl2 (Sigma-Aldrich) during 24 h, filtered, mechanically stirred with NaOH pellets (Synth, 40 g per 1 L EtOAc), filtered again and then distilled from P2O5 under inert atmosphere. Diisopropylamine (Aldrich) was refluxed over CaH2 (Aldrich) and distilled under an argon atmosphere. n-BuLi (1.6 mol L-1) in hexanes (Acros) was titrated with t-butanol/1,10-phenanthroline prior to use.57 Isobutyric acid and cyclopentanecarboxylic acid were refluxed and distilled from KMnO4, then refluxed and distilled from P2O5 and stored under argon. Trimethyl phosphite (Acros) was refluxed over sodium, decanted and distilled under argon prior to use. Methyl iodide (Acros) was distilled and stored at 4 ºC in the dark. 9,10-Phenanthrenequinone (Sigma-Aldrich) was recrystallized from 1,4-dioxane (mp 206-207 ºC, literature 206-207 ºC). 58,59N,N'- Dicyclohexylcarbodiimide (DCC, Acros) was used as received.
UV-Vis spectrophotometry
UV-Vis spectra were obtained with a Varian Cary 50 spectrophotometer with a cell holder thermostatized at 25.0 ± 0.5 ºC by a Varian Cary PCB 150 water-circulating bath. Peroxide concentrations were determined by iodometry (I3 absorption, ε353 = 2.55 × 104 L mol1 cm1),60 using an absorption cuvette with 3.0 mL of a 0.05 mol L1 potassium iodide solution in 0.1 mol L1 HOAc/OAc buffer (pH 3.8), containing 10 µL of a 1 mg mL1 aqueous solution of HRP-VI (Sigma, hydrogen-peroxidase oxidoreductase, EC 1.11.1.7, type VI-A, from horseradish) and 10 µL of a diluted peroxide solution in MeOH, in order to obtain an absorbance between 0.5 and 0.8 at 353 nm.
NMR spectroscopy
A Bruker AC200 (200 MHz) spectrometer was used (25 ºC, CDCl3) to obtain the spectra of non-peroxidic compounds. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard. For low temperature (< 10 ºC) peroxide characterization, NMR spectra were obtained on two Bruker spectrometers, DPX300 (300 MHz) and DRX500 (500 MHz), both equipped with low temperature probes.
Mass spectrometry
Low-resolution spectra (LR-MS) were obtained with a gas chromatographer coupled to a mass spectrometer GC-MS Shimadzu 14B/QP5050A with a quadrupole analyzer. A Zebron ZB-5 (30 m × 0.25 mm × 0.25 µm) column with split, helium as carrier gas and 70 eV as ionization energy were used. Injector temperature was at 250 ºC, oven temperature at 60 ºC (0 to 1 min) increasing 10 ºC min1 until 280 ºC and keeping this temperature constant for 33 min.
Infrared spectroscopy
Spectra were obtained on a FTIR Bomem MB100 spectrometer, operating between 4000 and 350 cm1.
Elemental analysis
The CHN composition of samples was obtained in a Perkin-Elmer CHN 2400 analyzer. Benzoic acid was used as a standard, resulting in measurements with a standard deviation (sd) of 0.3%.
TLC at low temperature
Despite the extreme instability of 1,2-dioxetanone derivatives, it was possible to perform TLC (thin layer chromatography) analysis for derivatives 5 and 6 using a special methodology; this analysis was used to follow reaction progression. The TLC plates (Merck, 2 × 5 cm Kieselgel 60 F254 over aluminum foil) were eluted in a closed glass chamber, which had been previously placed in a thermally insulated box containing small chunks of dry ice. Using this simple procedure, it was possible to elute samples of 5 and 6 at low temperature and detect the peroxidic spots by development with aqueous 10% KI solution. However, 1,2-dioxetanone 4 proved to be too unstable to be analyzed by TLC even with this methodology.
Synthesis of cyclic peroxides
Caution! Crystals of organic peroxides tend to explode even with minor impacts and are extremely sensitive to temperature increase! Careful handling is recommended during the preparation and isolation of such compounds!
Diphenoyl peroxide (3)
Trimethyl phosphite (0.75 mL, 6.4 mmol) was added slowly under stirring to a pale yellow suspension of 9,10-phenanthrenequinone (1.2 g, 5.8 mmol) in 60 mL dry toluene. After 30 min, the suspension became a slightly orange clear solution, which was kept stirring at room temperature for another 6 h under an argon atmosphere. The solvent was removed at low pressure, yielding a brownish oil, which was dissolved in 3.0 mL of dry hexane. The mixture was cooled in an ice/water bath, affording crystals of the desired phosphorane adduct. The solvent was carefully removed with a pipette and the crystals dried under an inert gas flow.
For the ozonization, the obtained phosphorane crystals were dissolved in dry CH2Cl2 and transferred to a round-bottomed flask fitted with a cannula. Using an acetone/dry ice slush bath, the solution was cooled to 78 ºC, and ozone was gently bubbled through the mixture for 6 h, using an Aqua Zone ozonisator (Red Sea Fish pHarm Ltd.) at maximum power, producing ca. 0.1 mmol min1 of ozone. After this period, the solvent was removed at low pressure and temperatures below 10 ºC; 30 mL of MeOH were added and the mixture was cooled to 70 ºC for 40 min, allowing the crystallization of the produced peroxide. The supernatant was removed with a pipette under N2 atmosphere, 2.0 mL of CH2Cl2 and 3.5 mL of MeOH were then added, and the mixture allowed to rest for 1 h in an ice/water bath. After complete crystallization, the supernatant was removed carefully with a pipette and under nitrogen flow. The solid was kept for 10 min under high vacuum (< 1 mmHg) at 0 ºC, affording 310 mg (22%) of 3 as colorless needles, which were stored at 20 ºC.
IR (KBr) ν/cm-1 1758, 1281, 1230, 1065, 1012, 771; anal. found (calc.) % for C14H8O4 C 69.45 (70.00), H 2.91 (3.36), N 0.50 (0.00); 1H NMR (500 MHz, CDCl3, 10 ºC) δ 7.37 (ddd, 2H, Jorto 7.7 Hz, Jmeta 0.7 Hz, H3), 7.62 (dd, 2H, Jorto 7.6 Hz, Jmeta 1.2 Hz, H2), 7.69 (ddd, 2H, Jorto 7.6 Hz, Jmeta 1.4 Hz, H4), 7.76 (dd, 2H, Jorto 7.5 Hz, Jmeta 1.1 Hz, H5); 13C NMR (125 MHz, CDCl3, 10 ºC) δ 128.0 (C5), 128.3 (C3), 129.4 (C1), 130.6 (C2), 132.9 (C4), 136.0 (C6), 171.3 (C=O).
2-Hydroperoxy-2-methylpropanoic acid (7)
In a 250 mL vacuum-flame dried triple-necked round-bottomed flask, 4.2 mL (30 mmol) of dry diisopropylamine and 70 mL of dry THF were mixed under inert atmosphere. An acetone/dry ice slush bath was used to keep the temperature between 60 and 40 ºC while, under vigorous stirring, 20 mL (31.5 mmol, 1.05 eq.) of 1.6 mol L-1n-BuLi solution in hexanes were slowly added with a syringe. After addition, the bath was removed, the flask was allowed to warm up slowly to room temperature, and stirred for additional 10 min; then the flask was cooled again to 78 ºC and 1.2 mL (12.5 mmol) of anhydrous isobutyric acid diluted in 5.0 mL of dry THF were added through a syringe. The resulting mixture was allowed to reach room temperature and then heated at 50 ºC for 1 h, obtaining a clear yellow solution, characteristic for the presence of a dianion. The solvent and the diisopropylamine were removed under vacuum (room temperature, 5 mm Hg) and the obtained white solid dissolved in 60 mL of dry THF. Additionally, a second triple-necked round-bottomed flask, equipped with septum, mechanical stirring (sealed with vacuum grease) and a system of nitrogen flow, was charged with 70 mL of dry THF and its temperature lowered to < 70 ºC using a liquid nitrogen/ethanol slush bath. The solvent in this flask was saturated with dry oxygen gas by bubbling with a needle for 10 min. The dianion solution in the first flask was then slowly transferred to the oxygen-saturated solution in the second flask using a cannula, while keeping the oxygenation flask at low temperature and under strong oxygen flow and mechanical stirring. After the dianion addition was completed, the mixture was left stirring for 2 h. Still under strong stirring and with cooling, 5.0 mL of a 36% aqueous HCl solution were added, and the mixture was left stirring for another 30 min, allowed to warm up to 20 ºC and transferred to a 500 mL separatory funnel containing 100 mL of a cold saturated NaCl solution. The aqueous layer was extracted with 5 × 25 mL Et2O and 5 × 25 mL CH2Cl2, while keeping the temperature of the solution the lowest possible by the addition of ice chunks. The combined organic layers were dried with MgSO4 at 4 ºC for 10 min, filtered at 0 ºC and stored at 20 ºC. The crude product was concentrated by evaporation under reduced pressure, keeping the bath at 0 ºC, to obtain a yellow peroxidic oil (positive peroxide test with 10% aqueous KI solution). The oil was purified by recrystallization at low temperature under inert atmosphere from Et2O/pentane by dissolving it between 0 and 5 ºC and crystallizing at 20 ºC, obtaining colorless crystals. The supernatant solvent was removed using a Pasteur pipette with a special very narrow tip. The remaining traces of solvent were removed under vacuum (< 1 mmHg) at 30 ºC to finally obtain 690 mg (46% yield) of 7.
Rf = 0.2 (Hex/EtOAc 1:1), 0 (CH2Cl2) and 0.3 (EtOAc); IR (CHCl3) ν/cm-1 3620, 3450, 1715; 1H NMR (500 MHz, CDCl3, 0 ºC) δ 1.51 (s, 6H, two CH3), 9.51 (bs, 2H, COOH and -OOH); 13C NMR (125 MHz, CDCl3, 0 ºC) δ 22.4 (CH3, C3), 83.5 (C(CH3)2, C2), 180.3 (C=O).
2-Carboxy-2-hydroperoxyadamantane (8)
The overall procedure is similar to the one used for 7. A 250 mL vacuum-flame dried triple-necked round-bottomed flask was charged with 5.6 mL (40 mmol) of dry diisopropylamine and 45 mL of dry THF, under a strong argon flow. The system was then cooled with an acetone/dry ice slush bath at 78 ºC and 27 mL (42 mmol) of a 1.54 mol L-1n-BuLi solution in hexanes were slowly added through a syringe. The temperature was raised to 0 ºC, kept constant for 20 min, and then lowered again to 20 ºC. Using a syringe, 3.0 g (17 mmol) of 2-adamantanecarboxylic acid (see its preparation in the Supplementary Information section) dissolved in 20 mL of dry THF were slowly added and the solution allowed to reach room temperature under stirring. The solvent and the amine were removed under vacuum (room temperature, 5 mm Hg), the obtained white solid dissolved in 200 mL of dry THF and slowly transferred during 3 h, through a cannula, to another round-bottomed flask already charged with 150 mL of dry THF saturated with oxygen gas and stirred vigorously, placed in a dry ice/acetone slush bath at 78 ºC. After one additional hour of stirring at 78 ºC, 30 mL of a 10% aqueous solution of HCl were added still under argon atmosphere. The temperature was allowed to rise to 20 ºC and the reaction mixture extracted with Et2O (3 × 50 mL). The combined organic layers were dried with MgSO4 for 10 min at 4 ºC, filtered and concentrated under vacuum in a water/ice bath. The crude peroxidic product was purified by column chromatography at 45 ºC, using petroleum ether (30-70 ºC)/Et2O 1:1 as eluent. The peroxide 8 (710 mg, 20%) was obtained as a colorless solid.
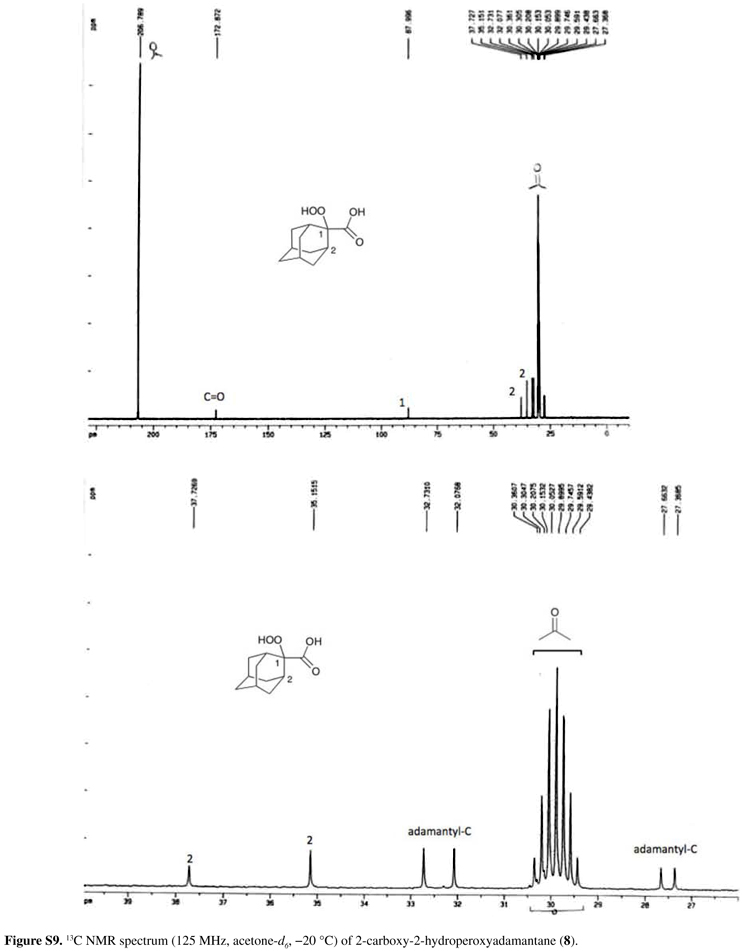
Rf = 0.3 (petroleum ether/Et2O 1:1); IR (KBr) ν/cm-1 3600-2300, 3429, 2932, 2861, 1691, 1454, 1296, 1274, 1104, 1065; anal. found (calc.) % for C11H16O4 C 63.13 (62.25), H 7.34 (7.60),; 1H NMR (500 MHz, acetone-d6, 20 ºC) δ 1.45-2.16 (m, 12H, adamantyl-H), 2.39 (bs, 2H, adamantyl-H, H2), 11.0 (bs, 1H, OOH), 11.2 (bs, 1H, COOH); 13C NMR (125 MHz, acetone-d6, 20 ºC) δ 27.4, 27.7, 32.1, 32.7, 35.2 (C2), 37.7 (C2), 88.0 (C1), 172.9 (C=O).
1-Carboxy-1-hydroperoxycyclopentane (9)
The overall procedure is similar to the one used for 7. A 250 mL vacuum-flame dried triple-necked round-bottomed flask was charged with 4.2 mL (30 mmol) of dry diisopropylamine and 70 mL of dry THF under a strong argon flow. The system was then cooled with an ethanol/dry ice slush bath at 45 ºC, and 20 mL (32 mmol) of 1.6 mol L-1n-BuLi solution in hexanes were slowly added through a syringe. After 15 min, the reaction mixture was allowed to reach room temperature, left stirring for 25 min, cooled to 78 ºC, and 1.4 mL (12.5 mmol) of freshly distilled cyclopentanecarboxylic acid dissolved in 5.0 mL of dry THF were slowly added. After 10 min, the temperature was allowed to rise to room temperature and the reaction mixture left stirring for more 2 h. The solvent and the amine were removed under vacuum (room temperature, 5 mmHg) and the obtained white solid dissolved in 60 mL of dry THF and slowly transferred, through a cannula, to another round-bottomed flask already charged with 100 mL of dry THF saturated with oxygen gas and vigorously stirred, placed in a dry ice/ethanol slush bath at 78 ºC. The transfer was performed within 2 h and the reaction was carried on for another 2 h, while oxygen was bubbled vigorously in the reaction flask. After complete reaction, 30 mL of a 10% aqueous solution of HCl were added at 78 ºC, the mixture stirred for more 30 min and the temperature allowed to rise to 20 ºC. The solution was then transferred to a 500 mL separatory funnel containing 200 mL of a cold saturated NaCl solution and the aqueous layer extracted with 5 × 50 mL Et2O, keeping the temperature low by adding ice chunks. The combined organic layers were dried over MgSO4 at 4 ºC for 10 min, filtered at 0 ºC and concentrated by evaporation under reduced pressure, keeping the bath temperature always below 2 ºC, obtaining a slightly yellow solid. This solid was dissolved at 0 ºC in pentane and allowed to crystallize at 20 ºC under an inert gas flow. The supernatant was removed with a Pasteur pipette and the clear yellow cubic crystals were dried under vacuum (< 1 mmHg) at 30 ºC to obtain 970 mg (53% yield) of 9.
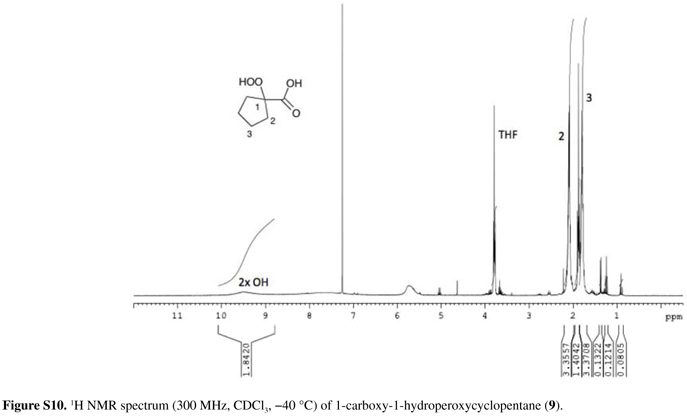
Rf = 0.4 (Hex/EtOAc 1:1) and 0.0 (CH2Cl2); 1H NMR (300 MHz, CDCl3, 40 ºC) δ 1.67-1.91 (m, 4H, H3), 1.98-2.21 (m, 4H, H2), 9.51 (bs, 2H, COOH and OOH); 13C NMR (75 MHz, CDCl3, 40 ºC, Figure S11) δ 25.1 (C3), 35.3 (C2), 93.4 (C1), 180.6 (C=O).
3,3-Dimethyl-1,2-dioxetanone (4)
A 250 mL triple-necked round-bottomed flask (flask one), equipped with septum and magnetic stirring, was connected to a 100 mL twin-necked round-bottomed flask (flask two) through a U-shaped glass tube in order to perform a bulb-to-bulb distillation. To avoid the transfer of solid particles between flasks during distillation, a bit of glass wool was placed inside the glass tube. The whole set-up was vacuum-flame dried and then filled with argon. Through a syringe, 0.75 g (6.3 mmol) of 7 dissolved in 2.0 mL of dry CH2Cl2 was added to flask one, the temperature was lowered to 78 ºC with an acetone/dry ice slush bath and 6.5 mL (6.5 mmol) of a 1.0 mol L-1 DCC solution in CH2Cl2 were added. After 15 min of stirring, additional 0.5 mL of the DCC solution was added and the solution was left stirring for another 30 min. Flask two was placed within a liquid nitrogen bath and the α-peroxylactone 4, together with the solvent, was vacuum distilled (1 mmHg) from reaction flask one to flask two. Flask one was kept at 30 ºC, under strong stirring, during the distillation, which was continued until a dry white paste was obtained in flask one, when 1.5 mL of CH2Cl2 were added and distillation was continued. Six successive additions of CH2Cl2 and distillations were realized during a 2 h period. The peroxidic solution of 4, with concentration of (1.7 ± 0.5) × 10-2 mol L-1 (5% yield), was stored in several portions in separate vials, kept at 80 ºC.
For the NMR analysis, the synthesis of 4 was realized directly in CDCl3. The general procedure described above was followed, with minor modifications. In this preparation, 260 mg (2.2 mmol) of 7 were dissolved in 1 mL of CDCl3 (previously treated with K2CO3) and added to the reaction flask at 40 ºC, followed by 450 mg (2.2 mmol) of DCC dissolved in a minimal quantity of CDCl3 (< 1 mL). After 15 min stirring, the mixture was subjected to bulb-to-bulb distillation as described before until all solvent was removed from flask one. In order to obtain a maximum concentration of 4 in CDCl3 solution, only one distillation was performed.
1H NMR (500 MHz, CDCl3, 20 ºC) δ 1.80 (s, 6H, two CH3); 13C NMR (125 MHz, CDCl3, 20 ºC) δ 22.1 (CH3, C2), 99.0 (C(CH3)2, C1), 169.8 (C=O).
spiro-Adamantyl-1,2-dioxetanone (5)
A vacuum-flame dried twin-necked round-bottomed flask was charged with 231 mg (1.09 mmol) of 8 dissolved in 50 mL of CH2Cl2, while keeping the temperature at 40 ºC using an acetone/dry ice slush bath, and 225 mg (1.09 mmol) of DCC dissolved in 4.3 mL of CH2Cl2 were slowly added. The reaction mixture was strongly stirred during 5 h at 40 ºC, while being monitored by TLC analysis. The insoluble urea derivative generated during the reaction was removed by filtering the mixture on a column packed with florisil (3 g) at 45 ºC. The solvent was then evaporated under reduced pressure at a temperature not higher than 20 ºC, yielding 120 mg (55%) of a slightly yellow solid. For further purification, the product was recrystallized three times from n-pentane between 25 ºC (dissolution) and 50 ºC (crystallization) giving pure 8 as slightly yellow cubic crystals, in a total yield of less than 10%.
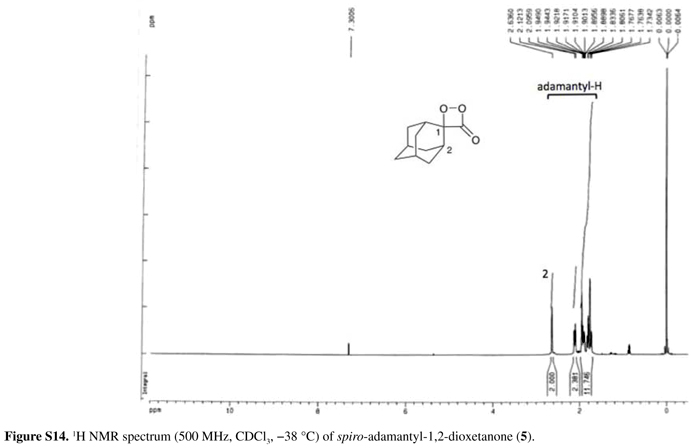
Rf = 0.7 (Hex/EtOAc 1:1); 1H NMR (500 MHz, CDCl3, 38 ºC) δ 1.71-1.97 (m, 10H, adamantane-H), 2.07-2.10 (m, 2H, adamantane-H), 2.61 (bs, 2H, adamantane-H, H2); 13C NMR (125 MHz, CDCl3, 38 ºC) δ 25.2, 25.3, 31.7, 33.0, 33.5 (C2), 35.3 (C2), 104.6 (C-spiro-Ad, C1), 169.3 (C=O).
spiro-Cyclopentyl-1,2-dioxetanone (6)
The overall procedure is similar to the one used for 4, connecting two round-bottomed flasks through a U-shaped glass tube. Flask one was charged with 413 mg (2.8 mmol) of 9 dissolved in 3.0 mL of cold CH2Cl2, while keeping the temperature at 30 ºC with an ethanol/dry ice slush bath, and 580 mg (2.8 mmol) of DCC, dissolved in 3.0 mL of CH2Cl2, were slowly added. After 2 h, the reaction was completed, as verified by TLC analysis at low temperature, using CH2Cl2 as eluent: Rf = 0.7 (6), 0 (9). While keeping the temperature of flask one at 30 ºC and flask two immersed in liquid nitrogen, the α-peroxylactone 6 was isolated through bulb-to-bulb distillation (< 1 mmHg). After distillation until dryness in flask one, additional 1.5 mL of CH2Cl2 were added and distillation was resumed. Five cycles of CH2Cl2 addition and distillation were performed during a 2 h period. The peroxidic solution of 6, with a concentration of (2.4 ± 0.1) × 10-3 mol L-1 (1.2% yield), was stored in several vials kept at 80 ºC.
For the NMR analysis, the synthesis of 6 was realized directly in CDCl3, as done for 4. The general procedure described above was followed, with minor modifications. Addition of 290 mg (1.4 mmol) of DCC dissolved in a minimal quantity of CDCl3 (< 1 mL) to 200 mg (1.4 mmol) of 9 at 30 ºC in flask one was followed by 15 min stirring and bulb-to-bulb distillation until dryness. Only one distillation was performed to obtain a high concentration of 6 (4.1 mmol L-1) in CDCl3.
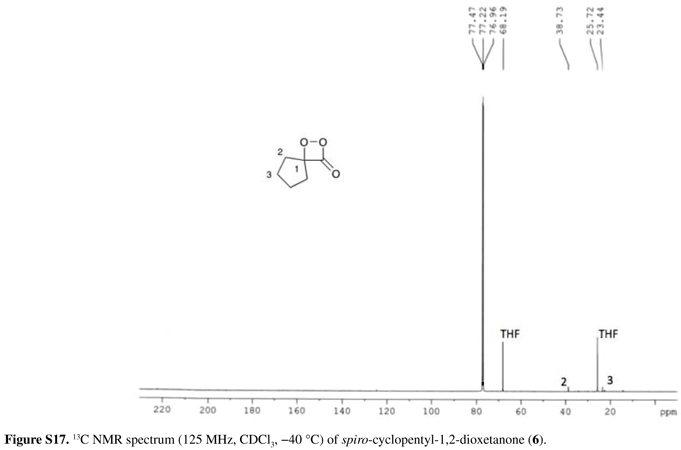
1H NMR (500 MHz, CDCl3, 40 ºC) δ 1.89-1.94 (m, 4H, H3), 2.00-2.01 (m, 2H, H2), 2.12-2.15 (m, 2H, H2); 13C NMR (125 MHz, CDCl3, 40 ºC) δ 23.4 (C3), 38.7 (C2), both with low intensities. No signal at 220.6 ppm, corresponding to the cyclopentanone carbonyl carbon, the peroxide decomposition product, was observed.
Results and Discussion
Diphenoyl peroxide (3)
The first report on the synthesis of diphenoyl peroxide (3) describes a two-step one-pot procedure based on the preparation of the trimethylphosphite adduct of 9,10-phenanthrenequinone followed by ozonization (Scheme 3).61,62 The first step is carried out in toluene, followed by ozone bubbling for 30 min, and the peroxide 3 is recrystallized from MeOH/CH2Cl2 1:1. After reproducing this method, the group concluded that its major drawback is the presence of the starting material after recrystallization of 3, as determined by TLC analysis.
Consequently, it was used a slightly modified procedure to obtain 3. It consists in the reaction of 9,10-phenanthrenequinone and trimethylphosphite in toluene until complete consumption of the quinone (after about 8 h, the reaction mixture becomes clear indicating complete consumption of the quinone, insoluble in toluene).62 The unstable adduct was then recrystallized from hexane (yield not determined) and immediately submitted to the ozonization reaction in toluene.
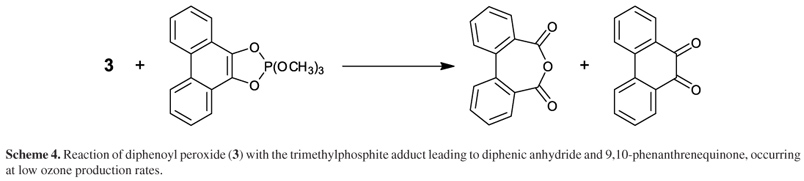
In a representative preparation, the ozonization product afforded 310 mg (22%) of 3 as colorless needles, after three recrystallizations from MeOH/CH2Cl2. High ozone production rates (> 0.1 mmol min1) were needed to increase the peroxide formation yield (> 10%) since in low ozone concentrations the reaction between 3 and the quinone-trimethylphosphite adduct becomes significant, leading to the starting 9,10-phenanthrenequinone and diphenic anhydride (dibenzo[c,e]oxepine-5,7-dione) as side products (Scheme 4).62
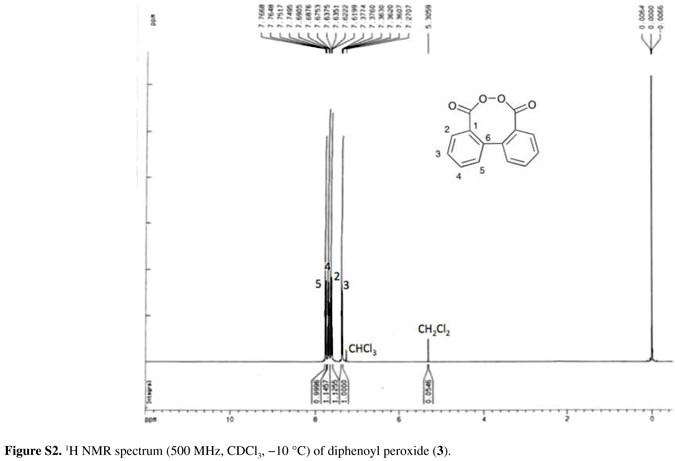
Dried crystals of 3 are extremely sensitive to even minor impacts, and must be handled with care. Herein, it is reported for the first time the 1H and 13C NMR spectra of 3, obtained in CDCl3 at 10 ºC on a 500 MHz spectrometer, completing its characterization together with the IR absorption frequencies and elemental analysis (see the Experimental section). Until now, the characterization of 3 had been performed solely based on elemental analysis and other indirect evidences.62 The carbonyl group appeared at 1758 cm1 in the IR spectrum and the carbonyl carbon at 171.3 ppm in the 13C NMR spectra. No evidences were found for the presence of starting materials, diphenic anhydride or benzocoumarin, a possible product formed in the thermolysis of 3 (Scheme 5).27
1,2-Dioxetanones (4-6)
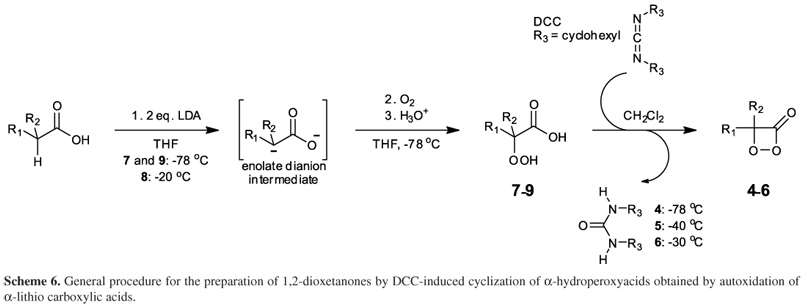
1,2-Dioxetanones 4-6 were prepared according to a general literature procedure,63-67 introducing specific modifications for each of the derivatives (see the Experimental section for details). The methodology consists in cyclization of an α-hydroperoxyacid derivative (7-9) with N,N'-dicyclohexylcarbodiimide (DCC), in which 7-9 are obtained by α-lithiation of the corresponding carboxylic acids, followed by low-temperature autoxidation (Scheme 6).
The preparation of the α-hydroperoxyacids is extremely difficult as it requires low temperatures, anhydrous conditions (in order to produce the enolate dianion) and a reaction medium free of transition metals (to avoid redox-induced peroxide decomposition). To avoid transition metal contamination, all glassware was thoroughly washed with aqueous EDTA solutions and EDTA-purified CH2Cl2 prior to use. Additionally, α-hydroperoxyacids are highly hygroscopic, thermally unstable and tend to undergo acid and base-catalyzed decarboxylation via Grob fragmentation.68 The preparation of 2-hydroperoxy-2-methylpropanoic acid (7)65 and 2-carboxy-2-hydroperoxyadamantane (8)67 had already been reported before; however, 1-carboxy- 1 hydroperoxycyclopentane (9) is a novel derivative.
After formation of the enolate dianion from the corresponding carboxylic acid derivative by addition of two equivalents of LDA at low temperature, the diisopropylamine formed from LDA was removed in vacuum (room temperature, 5 mmHg) together with the solvent THF. Removal of the amine before oxygenation is essential for the success of the preparation, to avoid electron transfer catalyzed decomposition of the peroxide by the electron rich amine.26,38 Attempts to obtain 7-9 directly using n-BuLi as base instead of LDA were not successful. Additionally, for an efficient preparation of α-hydroperoxyacids, it is necessary to slowly add the enolate dianion dissolved in THF to oxygen saturated THF at about 70 ºC, not the contrary. Oxygen bubbling into a solution of the dianion at low temperature does not lead to α-hydroperoxyacid formation. The α-hydroperoxyacid derivatives can be isolated by aqueous work-up at low temperature and fast low-temperature extraction. For derivatives 7 and 9, the crude products were purified by low-temperature recrystallization from diethylether/pentane mixtures to provide colorless or yellowish crystals, in 46 and 53% yield, respectively. However, derivative 8 proved to be stable enough for column chromatographic purification at 45 ºC and it was obtained in pure form as colorless solid in 20% yield. Interestingly, when more than 200 mg of crude 8 were applied to the chromatographic column, much lower product yields were obtained. The α-hydroperoxyacids were characterized by their low-temperature 1H and 13C NMR spectra as well as their infrared spectra (except 9), which are in agreement with the proposed structures (see the Experimental section).
The 1,2-dioxetanone derivatives 4-6 were obtained by cyclization/dehydratation of the corresponding α-hydroperoxyacids activated by DCC (Scheme 6). The reaction sequence is initiated by proton transfer from the carboxylic acid to DCC followed by nucleophilic attack of the carboxylate group onto the protonated DCC molecule.69 Intramolecular proton transfer from the α-hydroperoxy group to the DCC nitrogen atom transforms this moiety into an excellent leaving group, allowing the cyclization reaction by intramolecular nucleophilic addition of the peroxy anion onto the derivatized carboxyl carbon followed by the liberation of N,N'-dicyclohexylurea and 1,2-dioxetanone formation. At the low temperatures at which this reaction takes place, the resulting urea derivative is insoluble in CH2Cl2, facilitating product isolation and inhibiting peroxide decomposition (Scheme 7).
1,2-Dioxetanone 4 was synthesized by cyclization of 7 with DCC at 78 ºC and a total reaction time of 45 min, whereas 5 was similarly prepared from 8 at 40 ºC in 5 h and the derivative 6 obtained from 9 at 30 ºC with a 2 h reaction time. Reaction progress was always accompanied by TLC analysis (see Experimental section). The isolation of 1,2-dioxetanones must occur immediately after their preparation, given their susceptibility to decompose in the reaction media. Derivatives 4 and 6 were isolated together with the solvent by vacuum (1 mmHg) bulb-to-bulb distillation with the reaction flask at 30 ºC and the recipient flask at 78 ºC, resulting in peroxidic solutions with 17.0 and 2.4 mmol L1 concentration of 4 and 6, corresponding to yields of 5 and 1.2%, respectively. The lower peroxide concentration obtained for 6 might be due to a somewhat lower volatility of this compound when compared to 4. Stock solutions of these two 1,2-dioxetanones in CH2Cl2 could be more conveniently stored when maintained in several 1.5 mL glass vials kept at 80 ºC, which were defrosted and opened just before use in kinetic experiments.39
1,2-Dioxetanone 5 could not be isolated through bulb-to-bulb distillation due to its much lower volatility, therefore, the reaction mixture was filtered over florisil at 45 ºC to remove the urea derivative and the solvent evaporated at temperature less than 20 ºC, leading to a slightly yellow solid in 55% yield. This peroxide could be further purified by low temperature recrystallization from n-pentane, being obtained as slightly yellow cubic crystals (< 10% yields).
Peroxides 5 and 6 could be analyzed by TLC using a specially developed low-temperature method (see the Experimental section for details); however, derivative 4 proved to be too instable for analysis even using this methodology. For the NMR analysis at low temperature, the preparation of 4 and 6 was carried out in anhydrous CDCl3 (without TMS for 6) directly prior to spectra acquisition, assuring the highest possible peroxide concentration and low contamination by decomposition products. The 1H NMR spectrum of 4 showed one singlet at 1.80 ppm for the two methyl groups; after heating the NMR tube at 25 ºC for 1 h, a new strong singlet at 2.22 ppm was observed, corresponding to the decomposition product acetone. As expected, the two methylene groups appeared as multiplets at 2.00 and 2.12 ppm in the 1H NMR spectrum of 6.
The 13C NMR spectrum of 4 showed the expected three signals: at 22.1 ppm for the two methyl groups, at 99.0 ppm for the saturated ring-carbon and at 169.8 ppm for the C=O carbon. Similarly, the 13C NMR spectrum of 5 showed a signal at 104.6 ppm for the spiro carbon and at 169.3 ppm for the carbonyl carbon, together with the carbon signals corresponding to the adamantyl moiety. The new 1,2-dioxetanone 6 could only be obtained in a maximum concentration of 4.1 mmol L1, therefore it was not possible to observe the spiro carbon and the C=O carbon in its 13C NMR spectrum. Unfortunately, attempts to obtain CDCl3 stock solutions of 6 with higher concentrations failed. The difficulties in detecting quaternary and carbonyl carbon atoms of unstable compounds had already been observed during the 13C NMR spectroscopic characterization of peroxalic acids,70 even though these peroxides could be obtained in much higher concentrations. Nonetheless, the signal at 220.6 ppm relative to the decomposition product cyclopentanone could not be observed in the 13C NMR spectrum of 6; however, this signal was observed after heating the NMR tube at 25 ºC for 1 h. This observation, together with the 1H NMR spectrum of 6, its TLC analysis, as well as its chemiluminescence behavior,39 clearly indicates the identity of this new 1,2-dioxetanone derivative.
Conclusion
In this study the synthesis, purification and characterization of four cyclic organic peroxides, the 1,2-dioxetanone derivatives 4-6 and diphenoyl peroxide (3), are reported. These peroxides are extremely unstable and it was given herein a detailed description of the procedures used for their synthesis and characterization, mainly by 1H and 13C NMR spectroscopy. The general preparation procedure established for the synthesis of 1,2-dioxetanone derivatives can be used for the synthesis of other derivatives of this class of cyclic peroxides, which are of extreme importance for mechanistic chemiluminescence research.
The compounds 3 to 6, whose synthesis is described here in details, have already been used by our research group to unequivocally demonstrate that the intermolecular CIEEL (chemically initiated electron exchange luminescence) decomposition of this kind of peroxides occurs with low quantum yields, indicating that they do not constitute adequate models for efficient bioluminescence transformations and that better model systems have to be designed, synthesized and studied in the future.39
Supplementary Information
Supplementary data for the preparation and characterization of 2-adamantanecarboxylic acid (precursor for 8), IR, 1H and 13C NMR spectra of compounds 3-9 (Figures S1 to S17) and pictures of critical steps in the preparation of 1,2-dioxetanones (Figures S18 to S22) are available free of charge at http://jbcs.sbq.org.br as a PDF file.
Acknowledgements
The authors thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, W. J. B. 2006/03420-7, F. H. B. 2005/58320-4, L. F. M. L. C. 2012/02428-5, E. L. B. 2007/00684-6 and 2011/23036-5), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, E. L. B. 304887/2010-2) and Coordenadoria de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) for financial support.
Submitted: August 21, 2012
Published online: December 7, 2012
FAPESP has sponsored the publication of this article.
Supplementary Information
The supplementary material is available in pdf: [Supplementary material]
- 1. Baader, W. J.; Stevani, C. V.; Bastos, E. L. In The Chemistry of Peroxides, vol. 2., ed. 1; Rappoport, Z.; ed.; Wiley & Sons: Chichester, UK, 2006, ch. 16.
- 2. Radziszewski, B.; Ber. Dtsch. Chem. Ges. 1877, 10, 70.
- 3. Albrecht, H. O.; Z. Phys. Chem. 1928, 136, 321.
- 4. Gleu, K.; Petsch, W.; Angew. Chem. 1935, 48, 57.
- 5. Chandross, E. A.; Tetrahedron Lett. 1963, 12, 761.
- 6. Staudinger, H.; Chem. Ber. 1925, 58, 1075.
- 7. Staudinger, H.; Dyckerhoff, K.; Klever, H. W.; Ruzicka, L.; Chem. Ber. 1925, 58, 1079.
- 8. Bastos, E. L.; Baader, W. J.; ARKIVOC 2007, 8, 257.
- 9. Liu, Y.-J.; De Vico, L.; Lindh, R.; J. Photochem. Photobiol., A 2008, 194, 261.
- 10. De Vico, L.; Liu, Y.-J.; Wisborg, K. J.; Lindh, R.; J. Phys. Chem. A 2007, 111, 8013.
- 11. Liu, F. Y.; Liu, Y.-J.; De Vico, L.; Lindh, R. A; J. Am. Chem. Soc. 2009, 131, 618.
- 12. Liu, F. Y.; Liu, Y.-J.; De Vico, L.; Lindh, R.; Chem. Phys. Lett. 2009, 484, 69.
- 13. Roca-Sanjuán, D.; Delcey, M. G.; Navizet, I.; Ferré, N.; Liu, Y.-J.; Lindh, R.; J. Chem. Theory Comput. 2011, 7, 4060.
- 14. Chen, S. F.; Liu, Y. J.; Navizet, I.; Ferré, N.; Fang, W. H.; Lindh, R.; J. Chem. Theory Comput. 2011, 7, 798.
- 15. Navizet, I.; Liu, Y.-J.; Ferré, N.; Xiao, H.-Y.; Fang, W.-H.; Lindh R.; J. Am. Chem. Soc. 2010, 132, 706.
- 16. Song, C.-I.; Rhee, Y. M.; Int. J. Quantum Chem. 2011, 111, 4091.
- 17. Song, C.-I.; Rhee, Y. M.; J. Am. Chem. Soc. 2011, 133, 12040.
- 18. Adam, W.; Baader, W. J.; J. Am. Chem. Soc. 1985, 107, 410.
- 19. Adam, W.; Baader, W. J.; Angew. Chem., Int. Ed. 1984, 23, 166.
- 20. Ciscato, L. F. M. L.; Bartoloni, F. H.; Weiss, D.; Beckert, R.; Baader, W. J.; J. Org. Chem. 2010, 75, 6574.
- 21. Ciscato, L. F. M. L.; Weiss, D.; Beckert, R.; Bastos, E. L.; Bartoloni, F. H.; Baader, W. J.; New J. Chem. 2011, 35, 773.
- 22. Kopecky, K. R.; Mumford, C.; Can. J. Chem. 1969, 47, 709.
- 23. Adam, W.; Liu, J.-C.; J. Am. Chem. Soc. 1972, 94, 2894.
- 24. Schuster, G. B.; Schmidt, S. P.; Adv. Phys. Org. Chem. 1982, 18, 187.
- 25. Turro, N. J.; Ramamurthy, V.; Scaiano, J. C.; Modern Molecular Photochemistry of Organic Molecules, 1st ed.; University Science Books: California, USA, 2010.
- 26. Adam, W.; Cilento, G.; Chemical and Biological Generation of Excited States, 1st ed.; Academic Press: New York, USA, 1982.
- 27. Schuster, G. B.; Acc. Chem. Res. 1979, 12, 366.
- 28. Koo, J.-Y.; Schuster, G. B.; J. Am. Chem. Soc. 1978, 100, 4496.
- 29. Schmidt, S. P.; Schuster, G. B.; J. Am. Chem. Soc. 1980, 102, 306.
- 30. Schmidt, S. P.; Schuster, G. B.; J. Am. Chem. Soc. 1978, 100, 1966.
- 31. Adam, W.; Simpson, A.; Yany, F.; J. Phys. Chem. 1974, 78, 2559.
- 32. Adam, W.; Cueto, O.; J. Am. Chem. Soc. 1979, 101, 6511.
- 34. Turro, N. J.; Chow, M.-F.; J. Am. Chem. Soc. 1980, 102, 5058.
- 34. Dixon, B. G.; Schuster, G. B.; J. Am. Chem. Soc. 1981, 103, 3068.
- 35. Dixon, B. G.; Schuster, G. B.; J. Am. Chem. Soc. 1979, 101, 3116.
- 36. Ando, Y.; Niwa, K.; Yamada, N.; Enomoto, T.; Irie, T.; Kubota, H.; Ohmiya, Y.; Akiyama, H.; Nat. Photonics 2008, 2, 44.
- 37. Koo, J.; Schmidt, S. P.; Schuster, G. B.; Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 30.
- 38. Catalani, L. H.; Wilson, T.; J. Am. Chem. Soc. 1989, 111, 2633.
- 39. de Oliveira, M. A.; Bartoloni, F. H.; Augusto, F. A.; Ciscato, L. F. M. L.; Bastos, E. L.; Baader, W. J.; J. Org. Chem., in press, DOI: 10.1021/jo301309v.
- 40. Lee, C.; Singer, L. A.; J. Am. Chem. Soc. 1980, 102, 3823.
- 41. Zaklika, K. A.; Kissel, T.; Thayer, A. L.; Burns, P. A.; Schaap, A. P.; Photochem. Photobiol. 1979, 30, 35.
- 42. Schaap, A. P.; Gagnon, S. D.; J. Am. Chem. Soc. 1982, 104, 3504.
- 44. Schaap, A. P.; Sandinson, M. D.; Handley, R. S.; Tetrahedron Lett. 1987, 28, 1155.
- 44. Schaap, A. P.; Handley, R. S.; Giri, B. P.; Tetrahedron Lett. 1987, 28, 1159.
- 45. Adam, W.; Bronstein, I.; Edwards, B.; Engel, T.; Reinhardt, D.; Schneider, F. W.; Trofimov, A. V.; Vasil'ev, R. F.; J. Am. Chem. Soc. 1996, 118, 10400.
- 46. Trofimov, A. V.; Mielke, K.; Vasil'ev, R. F.; Adam, W.; Photochem. Photobiol. 1996, 63, 463.
- 47. Nery, A. L. P.; Röpke, S.; Catalani, L. H.; Baader, W. J.; Tetrahedron Lett. 1999, 40, 2443.
- 48. Adam, W.; Bronstein, I.; Trofimov, A. V.; Vasil'ev, R. F.; J. Am. Chem. Soc. 1999, 121, 958.
- 49. Adam, W.; Trofimov, A. V.; J. Org. Chem. 2000, 65, 6474.
- 50. Adam, W.; Matsumoto, M.; Trofimov, A. V.; J. Am. Chem. Soc. 2000, 122, 8631.
- 51. Nery, A. L. P.; Weiss, D.; Catalani, L. H.; Baader, W. J.; Tetrahedron 2000, 56, 5317.
- 52. Matsumoto, M.; J. Photochem. Photobiol. C 2004, 5, 27.
- 53. Tanimura, M.; Watanabe, N.; Ijuin, H. K.; Matsumoto, M.; J. Org. Chem. 2010, 75, 3678.
- 54. Bastos, E. L.; Ciscato, L. F. M. L.; Weiss, D.; Beckert, R.; Baader, W. J.; Synthesis 2006, 11, 1781.
- 55. Ciscato, L. F. M. L.; Bastos, E. L.; Bartoloni, F. H.; Guenther, W.; Weiss, D.; Beckert, R.; Baader, W. J.; J. Braz. Chem. Soc. 2010, 21, 1896.
- 56. Ciscato, L. F. M. L.; Weiss, D.; Beckert, R.; Baader, W. J.; J. Photochem. Photobiol., A 2011, 218, 41.
- 57. Watson, S. C.; Eastham, J. F.; J. Organomet. Chem. 1967, 9, 165.
- 58. Helferich, B.; Chem. Ber. 1954, 87, 233.
- 59. Eistert, B.; Chem. Ber. 1964, 97, 2479.
- 60. Cotton, M. L.; Dunford, H. B.; Can. J. Chem. 1973, 51, 582.
- 61. Ramirez, F.; Desai, N. B.; Mitra, R. B.; J. Am. Chem. Soc. 1961, 83, 492.
- 62. Ramirez, F.; Bhatia, S. B.; Mitra, R. B.; Hamlet, Z.; Desai, N. B.; J. Am. Chem. Soc. 1964, 86, 4394.
- 63. Adam, W.; Blancafort, L.; J. Am. Chem. Soc. 1996, 118, 4778.
- 64. Adam, W.; Alzérreca, A.; Liu, J.-C.; Yany, F.; J. Am. Chem. Soc. 1977, 99, 5768.
- 65. Adam, W.; Cueto, O.; J. Org. Chem. 1977, 42, 38.
- 66. Adam, W.; Cueto, O.; Ehrig, V.; J. Org. Chem. 1976, 41, 370.
- 67. Adam, W.; Blancafort, L.; J. Org. Chem. 1997, 62, 1623.
- 68. Grob, C. A.; Schiess, P. W.; Angew. Chem., Int. Ed. 1967, 6, 1.
- 69. Adam, W. In The Chemistry of Functional Groups, Peroxides, 1st ed.; Patai, S.; ed.; John Wiley & Sons: New York, USA, 1983, ch. 24.
- 70. Stevani, C. V.; Campos, I. P. A.; Baader, W. J.; J. Chem. Soc., Perkin Trans. 2 1996, 1645.
Publication Dates
-
Publication in this collection
03 Jan 2013 -
Date of issue
Nov 2012
History
-
Received
21 Aug 2012 -
Accepted
07 Dec 2012