Abstracts
The current study describes that nor-β-lapachone and its arylamino derivatives, iodinated and methylated naphthoquinones and nor-β-lapachone-based 1,2,3-triazoles exhibited pronounced cytotoxic effects against four human leukemia cell lines (HL-60, K562, Molt-4 and Jurkat). Nor-β-lapachones arylamino substituted with potent activity were identified, revealing themselves as potential prototypes against tumor cell lines. Moreover, cells treated with these compounds showed DNA damage according to the standard comet assay, a finding that was, at least in part, due to increased intracellular levels of ROS. HL-60 cells were chosen to study the underlying molecular mechanisms of cytotoxicity. Drug-induced apoptosis in HL-60 cells was observed by flow cytometry analyses. Strains of Saccharomyces cerevisiae were used for a preliminary investigation into the mechanism of drug action on DNA topoisomerases. These results suggested that the cytotoxicity of these compounds apparently does not involve topoisomerase inhibition, but that treatment impairs DNA repair activity, thus triggering cell death. Considering their pro-oxidant properties, we investigated the ability of these compounds to induce apoptosis and chromosomal aberrations as micronuclei in Chinese hamster lung fibroblasts (V79 cells). Morphological apoptotic nuclei and micronuclei induction following drug treatment were observed, suggesting a correlation between DNA damage and apoptosis.
naphthoquinone; nor-β-lapachone; antileukemic activity; reactive oxygen species; genotoxicity; DNA repair
O presente estudo descreve a acentuada atividade citotóxica da nor-β-lapachona, seus derivados arilamino substituídos, naftoquinonas iodadas e metilada, além de nor-β-lapachonas 1,2,3-triazólicas, contra quatro linhagens de células de leucemia humana (HL-60, K562, Molt-4 e Jurkat). Nor-β-lapachonas arilamino substituídas foram identificadas com potente atividade, revelando-se como potenciais protótipos contra as linhagens tumorais descritas. Estudos utilizando o ensaio cometa evidenciaram danos ao ácido desoxirribonucleico (ADN) causado pelos derivados arilamino substituídos devido o aumento dos níveis intracelulares de espécies reativas de oxigênio (ERO's). Células de HL-60 foram selecionadas para a continuidade dos estudos de mecanismos moleculares subjacentes e apoptose induzida pelos derivados quinoidais foi observada por análise de citometria de fluxo. Cepas de Saccharomyces cerevisiae foram utilizadas para uma investigação preliminar sobre o mecanismo de ação em topoisomerases de ADN. Os estudos sugerem que, aparentemente, a citotoxidade dos compostos não envolve a inibição de topoisomerases, mas que o tratamento prejudica a atividade de reparação do ADN, provocando assim a morte celular. A capacidade em induzir apoptose e aberrações cromossômicas em fibroblastos de pulmão de hamster chinês (células V79) também foi investigada. Núcleos apoptóticos foram observados e nossos estudos indicam uma correlação entre dano ao ADN e apoptose.
ARTICLE
Potent antileukemic action of naphthoquinoidal compounds: evidence for an intrinsic death mechanism based on oxidative stress and inhibition of DNA repair
Bruno C. CavalcantiI,* * e-mail: nunim_br@hotmail.com, eufranio@ufmg.br ; Igor O. CabralI; Felipe A. R. RodriguesI; Francisco W. A. BarrosI; Danilo D. RochaI; Hemerson I. F. MagalhãesI; Dinara J. MouraII; Jenifer SaffiII; João A. P. HenriquesII; Tatiane S. C. CarvalhoIII; Manoel O. MoraesI; Cláudia PessoaI; Isadora M. M. de MeloIV; Eufrânio N. da Silva JúniorIV,* * e-mail: nunim_br@hotmail.com, eufranio@ufmg.br
INational Laboratory of Experimental Oncology, Department of Physiology and Pharmacology, Federal University of Ceará, 60430-270 Fortaleza-CE, Brazil
IIDepartment of Biophysics, Federal University of Rio Grande do Sul, 91501-970 Porto Alegre-RS, Brazil
IIINatural Products Research Nucleus, Federal University of Rio de Janeiro, 21941-971 Rio de Janeiro-RJ, Brazil
IVLaboratory of Synthetic and Heterocyclic Chemistry, Department of Chemistry, Institute of Exact Sciences, Federal University of Minas Gerais, 31270-901 Belo Horizonte-MG, Brazil
ABSTRACT
The current study describes that nor-β-lapachone and its arylamino derivatives, iodinated and methylated naphthoquinones and nor-β-lapachone-based 1,2,3-triazoles exhibited pronounced cytotoxic effects against four human leukemia cell lines (HL-60, K562, Molt-4 and Jurkat). Nor-β-lapachones arylamino substituted with potent activity were identified, revealing themselves as potential prototypes against tumor cell lines. Moreover, cells treated with these compounds showed DNA damage according to the standard comet assay, a finding that was, at least in part, due to increased intracellular levels of ROS. HL-60 cells were chosen to study the underlying molecular mechanisms of cytotoxicity. Drug-induced apoptosis in HL-60 cells was observed by flow cytometry analyses. Strains of Saccharomyces cerevisiae were used for a preliminary investigation into the mechanism of drug action on DNA topoisomerases. These results suggested that the cytotoxicity of these compounds apparently does not involve topoisomerase inhibition, but that treatment impairs DNA repair activity, thus triggering cell death. Considering their pro-oxidant properties, we investigated the ability of these compounds to induce apoptosis and chromosomal aberrations as micronuclei in Chinese hamster lung fibroblasts (V79 cells). Morphological apoptotic nuclei and micronuclei induction following drug treatment were observed, suggesting a correlation between DNA damage and apoptosis.
Keywords: naphthoquinone, nor-β-lapachone, antileukemic activity, reactive oxygen species, genotoxicity, DNA repair
RESUMO
O presente estudo descreve a acentuada atividade citotóxica da nor-β-lapachona, seus derivados arilamino substituídos, naftoquinonas iodadas e metilada, além de nor-β-lapachonas 1,2,3-triazólicas, contra quatro linhagens de células de leucemia humana (HL-60, K562, Molt-4 e Jurkat). Nor-β-lapachonas arilamino substituídas foram identificadas com potente atividade, revelando-se como potenciais protótipos contra as linhagens tumorais descritas. Estudos utilizando o ensaio cometa evidenciaram danos ao ácido desoxirribonucleico (ADN) causado pelos derivados arilamino substituídos devido o aumento dos níveis intracelulares de espécies reativas de oxigênio (ERO's). Células de HL-60 foram selecionadas para a continuidade dos estudos de mecanismos moleculares subjacentes e apoptose induzida pelos derivados quinoidais foi observada por análise de citometria de fluxo. Cepas de Saccharomyces cerevisiae foram utilizadas para uma investigação preliminar sobre o mecanismo de ação em topoisomerases de ADN. Os estudos sugerem que, aparentemente, a citotoxidade dos compostos não envolve a inibição de topoisomerases, mas que o tratamento prejudica a atividade de reparação do ADN, provocando assim a morte celular. A capacidade em induzir apoptose e aberrações cromossômicas em fibroblastos de pulmão de hamster chinês (células V79) também foi investigada. Núcleos apoptóticos foram observados e nossos estudos indicam uma correlação entre dano ao ADN e apoptose.
Introduction
Naphthoquinones are widely distributed in nature, and many clinically important antitumor drugs containing a quinone moiety (anthracyclines, mitomycin and mitoxantrones) have demonstrated anticancer activity.1,2 Studies of the antitumor properties and mechanisms of action of quinone derivatives have shown that they can act as topoisomerase inhibitors via DNA intercalation, and their toxicity can also be explained by oxidative stress with reactive oxygen species (ROS) generation, which are usually toxic to normal tissues, thus reducing the therapeutic utility of quinones.3-5
Despite recent technological advances, cancer treatment remains a challenge, and, in some circumstances, a complete remission is difficult to achieve. Currently, the investigation of natural products and chemical modifications to antitumor substances are among the most important strategies used in the search for new antineoplastic drugs.6 Among the cytotoxic naphthoquinones, β-lapachone has been the most extensively studied in recent years7 and is currently under multiple phase II clinical trials.8 In addition, the semisynthetic nor-β-lapachone and its derivatives have been the subject of several cytotoxicity studies in various human cancer cell lines.2 More recently, using molecular hybridization,9 our group synthesized a set of 1,2,3-triazole, 3-arylamino and 3-alkoxy-nor-β-lapachone derivatives and previously reported their elevated antiproliferative activity against human cancer cell lines.10,11 The strategy employed to synthesize the nor-β-lapachone derivatives was based on a modification over the C-ring moiety of the prototype β-lapachone. This approach has demonstrated the potential of this substance and became the base for the synthesis of new important antitumor compounds.
Continuing our program to develop naphthoquinones with anticancer activity, it was investigated the antileukemic activity of four 3-arylamino-nor-β-lapachone derivatives 1-4 (Scheme 1), a methylated and iodinated naphthoquinones 5-7 (Scheme 2) and nor-β-lapachone-based 1,2,3-triazoles 8-10 (Scheme 3), which manifested pronounced cytotoxicity against human cancer cells using several different human leukemia cell lines (HL-60, K562, Molt-4 and Jurkat).
Moreover, to further understand, the biological mechanism of the cytotoxic effect of more potent naphthoquinones, our group also investigated their effect on Saccharomyces cerevisiae strains deficient in topoisomerases. As naphthoquinones increase intracellular ROS production, the in vitro genotoxic effects of the test compounds in Chinese hamster lung fibroblasts (V79) cells were determined using the cytokinesis-block micronucleus test.
Experimental
Chemicals
Fetal bovine serum and Dulbecco's modified eagle medium (DMEM) were purchased from Cultilab (Campinas-SP, Brazil). RPMI 1640 medium, trypsin-EDTA, penicillin and streptomycin, low-melting point agarose and agarose were purchased from GIBCO® (Invitrogen, Carlsbad, CA, USA). Propidium iodide (PI), acridine orange (AO), ethidium bromide (EB), rhodamine 123 (Rho-123), cytochalasin-B (Cyt-B), glutathione reduced ethyl ester (GSH-OEt) and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Formamidopyrimidine DNA-glycosylase (FPG) and endonuclease III (ENDOIII) were obtained from New England BioLabs (USA). Doxorubicin (Doxolem® ) was purchased from Zodiac Produtos Farmacêuticos S/A (Brazil). All other chemicals and reagents used were of analytical grade.
The synthesis and isolation of arylamino-nor-β-lapachone derivatives 1-4 and nor-β-lapachone-based 1,2,3-triazoles 8-10 were previously described by our research group and the same methods were used to prepare the compounds herein used to perform the antitumoral studies.11,14 Compounds 5-7 were prepared as described by Pinto and co-workers.13 See Supplementary Information section for the NMR spectra of the compounds.
Lapachol (2-hydroxy-3-(3'-methyl-2'-butenyl)-1,4-naphthoquinone) was extracted from the heartwood of Tabebuia sp. (Tecoma) and purified by a series of recrystallizations in the appropriate solvent, generally hexane.10,11 The next step consisted of obtaining nor-lapachol from lapachol using Hooker's oxidation reaction.14 3-Bromo-nor-β-lapachone was prepared from nor-lapachol by a reaction with bromine and was then immediately reacted with the respective arylamine to yield the substituted nor-β-lapachones 1-4 (Scheme 1) in moderate to high yields as recently described by us.11
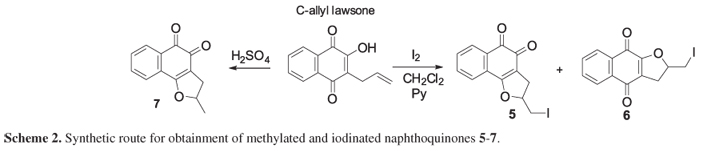
The methylated and iodinated compounds were obtained by methodology described by Pinto and co-workers13 from C-allyl lawsone. Initially, compounds 5 and 6 were synthesized from C-allyl lawsone by simple reaction with metallic iodine in dichloromethane. The substances 5 and 6 were obtained as crystalline solids (red and yellow crystals) after purification by column chromatography. Finally, the substance 7 was prepared by acid cyclization using sulfuric acid.13
The last class of evaluated compounds was prepared by methodology described by our research group.8Nor-β-lapachone-based 1,2,3-triazoles 8-10 were synthesized, as very recently described by us,12 in good yields, from 3-azido nor-β-lapachone by click chemistry reaction, methodology published by Sharpless and co-workers15 as shown in the Scheme 3.
Cell lines and cell cultures
The human myeloid leukemia cancer cell lines used in this report included the HL-60 (promyelogenous leukemia), K562 (chronic myelogenous leukemia), MOLT-4 (acute lymphoblastic leukemia) and Jurkat (T-cell lymphoblast leukemia) lines, which were obtained from the National Cancer Institute (Bethesda, MD, USA). Chinese hamster lung fibroblasts (V79 cells) were kindly provided by Dr. J. A. P. Henriques (Federal University of Rio Grande do Sul, Porto Alegre, Brazil).
Leukemia cells were maintained in RPMI 1640 medium, and V79 cells were cultivated under standard conditions in MEM with Earle's salts. All culture media were supplemented with 10% fetal bovine serum, 2 mmol L-1 glutamine, 100 µg mL-1 penicillin and 100 µg mL-1 streptomycin, and cultures were performed at 37 ºC with 5% CO2. To evaluate the cytotoxic and genotoxic activities of the tested compounds using V79 cells as a model, cells were grown for 2 days prior to treatment with the test substances, at which point, the medium was replaced with fresh medium containing the test substance or DMSO solution as a control. The final concentration of DMSO in the culture medium was kept constant, under 0.1% (v/v). All cell treatments were performed in triplicate and repeated at least 3 times.
Inhibition of tumor cell proliferation
The growth of the leukemia and V79 cell lines was quantified by the ability of living cells to reduce the yellow dye 3-(4,5-dimethyl-2-thiozolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) to a purple formazan product.16 For these experiments, myeloid leukemia and V79 cells were plated in 96 well plates (0.3 × 106 cells per well), and nor-β-lapachone and its derivatives 1-4 (0.02 to 13.07 µmol L-1), dissolved in DMSO (0.1%), were then added to each well and incubated for 72 h. In some experiments, the contribution of ROS to the cytotoxicity of some tested compounds (1-3) was measured by HL-60 cells pre-treated with a non-toxic concentration of GSH-OEt (15 mmol L-1) for 2 h. DMSO (0.1%) was used as a negative control. Subsequently, the plates were centrifuged, and the medium was replaced by fresh medium (150 µL) containing 0.5 mg mL-1 MTT. Three hours later, the MTT formazan product was dissolved in 150 µL DMSO, and the absorbance was measured using a multiplate reader (Spectra Count, Packard, Ontario, Canada). The drug effect was quantified as the percentage of control absorbance of the reduced dye at 595 nm.
Measurement of intracellular reactive oxygen species (ROS)
Levels of intracellular ROS were estimated following treatment with the various compounds using 2',7'-dichlorofluorescein diacetate (H2DCFDA) as a fluorescent probe. H2DCFDA readily diffuses through the cell membrane and is hydrolyzed by intracellular esterases to non-fluorescent dichlorofluorescein (DCFH), which is then rapidly oxidized to highly fluorescent 2',7'-dichlorofluorescein (DCF) by a broad range of intracellular oxidative stresses in addition to H2O2.17,18 Therefore, the increased mean fluorescence intensity of DCF can be used as a probe for a broad range of oxidative events not limited to H2O2. Briefly, leukemia cell lines were exposed to the test compounds (0.25, 0.5, 1 and 2 µmol mL-1) for 4 h, and the culture medium was then replaced by fresh serum-free medium containing 20 µmol mL-1 H2DCFDA. In another experimental set, HL-60 cells were pre-exposed to GSH-OEt (15 mmol L-1) for 2 h before treatment with some compounds (1-3). DCF fluorescence intensity was detected by flow cytometry using a Guava EasyCyteTM Mini (Guava Technologies, Inc., Hayward, CA, USA) and Guava Express Plus software. In general, the DCF fluorescence intensity is proportional to the amount of intracellular ROS.19
DNA strand breaks
The comet assay, which is used to detect DNA strand breaks, was conducted under alkaline conditions as described by Singh et al.20 with minor modifications21 and following the recommendations of the International Workshop on Genotoxicity Test Procedures.22 Leukemia cell lines were exposed to the test compounds (0.25, 0.5, 1 and 2 µmol L-1) for 4 h. Following exposure, slides containing treated cells for the comet assay were placed in the chilled lysis solution containing 2.5 mol L-1 NaCl, 100 mmol L-1 EDTA, 100 mmol L-1 Tris-HCl, 1% Trisma base, 1% Triton X-100 and 10% DMSO for 16 h at 4 ºC. The slides were then removed from the lysing solution and placed on a horizontal electrophoresis tank filled with freshly prepared alkaline buffer (300 mmol L-1 NaOH, 1 mmol L-1 EDTA, pH > 13.00). The slides were equilibrated in the same buffer for 20 min, and electrophoresis was performed at 25 V, 300 mA for 20 min. After electrophoresis, the slides were washed gently with 2 mol L-1 Tris-HCl buffer, pH 7.4, to remove the alkali. Each slide was stained with 50 µL ethidium bromide (20 µg mL-1), and a cover slip was placed on the slide. The analysis of the cells (100 cells from each of the three replicate slides) was performed using a visual scoring system23 that categorized tail length into five classes. The damage index, which is considered to be a sensitive DNA measure, was based on migration length as well as the amount of DNA in the tail. Therefore, a damage index value was assigned to each comet according to its class, and the values ranged from 0 (completely undamaged) to 400 (maximum damage).24 Doxorubicin (0.6 µmol L-1) was used as the positive control.25
Measurement of oxidized purines and pyrimidines
The alkaline comet assay was performed as described above in the HL-60 and K562 cell lines. At the end of the treatment (2 µmol L-1 for 4 h), the slides were removed from the lysing solution, washed 3 times in enzyme buffer (40 mmol L-1 HEPES, 100 mmol L-1 KCl, 0.5 mmol L-1 Na2EDTA, 0.2 mg mL-1 BSA, pH 8.0), and then drained and incubated with 70 µL FPG (30 min at 37 ºC) or ENDOIII (45 min at 37 ºC) diluted 1:103 in enzyme buffer according to manufacturer. In some experiments, the relevance of ROS on oxidative DNA damage (only oxidized purine bases by FPG-modified comet assay) of some tested compounds (1-3) was assessed by HL-60 cells pre-treated with GSH-OEt (15 mmol L-1) for 2 h. Images of 100 randomly selected cells (50 cells from each of two replicate slides) per group were visually analyzed. The amount of oxidized purines (FPG-sensitive sites) and pyrimidines (ENDOIII-sensitive sites) was then determined by subtracting the amount of strand breaks (samples incubated with buffer alone) from the total amount of breaks obtained after incubation with FPG and ENDOIII.
Saccharomyces cerevisiae strains and constructs
BY4741 haploid S. cerevisiae cells and two topoisomerase mutant isogenic strains (Table 1) were purchased from EUROSCARF (Institute of Microbiology, Johann Wolfgang Goethe University Frankfurt, Germany). The double mutant was constructed using a gpx3::URA3 disruption cassette that was amplified from YCPLac33 by PCR using specific primers [foward (5'ATGAAAGTGCTATGTGTCGCAGAGAAAAATT CTATAGCGAAATTAAATTGAAGCTCTAAT3') and reverse (5'TTACATGGATGCCTTGACACGGTCA TAAACTTGCAAGAGACATACTCTTCCTTTTT CAAT3')] and a high-fidelity thermostable DNA polymerase (Platinum® Taq, Invitrogen). The simple mutant top1Δwas transformed using the lithium acetate method26 with the linear disruption cassette gpx3::URA3, which generated the double mutant top1Δtop3Δ. Transformants were selected by growth on SynCo-Ura media, and disruption of the TOP3 gene was verified by genomic PCR.
Media and growth inhibition assay
Exponential phase cultures of S. cerevisiae strains were diluted in PBS (phosphate buffered saline; Na2HPO4, KH2PO4 and KCl; 20 mmol L-1; pH 7.4) to a density of 1 × 107 cells mL-1 for the growth inhibition assay. An inoculation loop of cells from a cell suspension was streaked from the center to the edge of a Petri dish in one continuous streak; test compounds at increasing concentrations (10, 20, 40 and 80 µg mL-1) were placed on a filter-paper disk in the center of the plate and incubated for 48 h at 30 ºC. Impaired growth was measured as mm of growth inhibition from the edge of the filter-paper disk to the beginning of cell growth. Values ranged from 0 (complete growth to the filter-disk) to 30 mm (absence of growth to the rim of the Petri dish). All tests were repeated at least three times for each treatment.
Analysis of mechanisms involved in cytotoxicity
The following experiments were performed to elucidate the mechanisms involved in the cytotoxic action of the active compounds in HL-60 cells, a well-established cell line suitable for examining the effects of test drugs on cell proliferation, cell cycle progression, cell differentiation, and apoptotic events.27,28 For the lipid peroxidation, nitrite/nitrate measurements and flow cytometric experiments described below, cells were exposed to increasing concentrations (0.25, 0.5, 1 and 2 µmol L-1) of test compounds for 4 h, and the cells were then reincubated for 20 h in the absence of the test substances.
Determination of lipid peroxidation (TBARS assay) and nitrite/nitrate production
Lipid peroxidation and the production of nitrite/nitrate as a result of nitric oxide (NO) release were measured in cell cultures using the standard methods described by Draper and Hadely29 and Green et al.30 All experiments were performed in triplicate in three independent experiments.
Determination of cell membrane integrity, internucleosomal DNA fragmentation, mitochondrial transmembrane potential (ΔΨm) and cell surface phosphatidylserine (PS)
Cell membrane integrity was evaluated by the exclusion of propidium iodide (PI) at 50 µg mL-1. To assay internucleosomal DNA fragmentation, the cells were incubated at 37 ºC for 30 min in the dark in a lysis solution containing 0.1% citrate, 0.1% Triton X-100 and 50 µg mL-1 PI, and the percentage of degraded DNA was determined according to the number of cells displaying subdiploid (sub-Go/G1) DNA in relation to the total number of cells examined.31
Transmembrane mitochondrial potential (ΔΨm) was evaluated using Rho-123 incorporation according to the method described by Marinho-Filho et al.32 Rho-123 is a cell-permeable, cationic, fluorescent dye that is readily sequestered by active mitochondria without inducing cytotoxic effects. Fluorescence was measured, and the percentage of mitochondrial depolarization was determined.
The annexin V flow cytometry assay was used to detect cell populations in apoptosis or necrosis, according to the method described Cavalcanti et al.33 The cells were stained with FITC-conjugated annexin V (Guava Nexin kit, Guava Technologies, Inc., Hayward, CA, USA) and PI (necrotic cell indicator), and they were then subjected to flow cytometric analysis.
For all cytometric experiments, cell fluorescence was determined by flow cytometry using a Guava EasyCyteTM Mini (Guava Technologies, Inc., Hayward, CA, USA) with Guava Express Plus software. 5,000 events were evaluated per experiment.
Kinetics of DNA repair and unscheduled DNA synthesis
To study DNA repair kinetics, HL-60 cells were treated with 8 × 10-5 mol L-1 methyl methanesulfonate (MMS) for 1 h and post-incubated in RPMI 1640 or post-treated in RPMI 1640 with 0.25 µmol L-1 of the naphthoquinones to be tested for 24 h. Cells not treated with MMS were also incubated in RPMI 1640 as a control. To measure DNA repair activity, the alkaline version of the comet assay was performed as described above.
Determination of unscheduled DNA synthesis (UDS), which principally measures global genome nucleotide excision repair (NER) activity, was performed as described by Bootsma et al.34 with minor modifications.31 UDS was assessed according to the incorporation of bromodeoxyuridine (BrdU) in the presence of hydroxyurea, a DNA synthesis inhibitor that does not inhibit repair mechanisms.35 In brief, HL-60 cells were treated for 24 h with the test compounds (5 µmol L-1) and were then washed twice with ice-cold PBS to remove the compounds. Then, the cells were immediately incubated for an additional 30 min with medium containing 3 mmol L-1 hydroxyurea, at which point, the culture medium was supplemented with BrdU (10 mmol L-1). After 2 h incubation, the cells were washed (PBS), harvested and transferred to cytospin slides, which were allowed to dry for 2 h at room temperature. Cells that had incorporated BrdU were labeled by direct peroxidase immunocytochemistry utilizing the chromogen diaminobenzidine. Slides were counterstained with hematoxylin, mounted, and covered with a cover slip. Evaluation of BrdU positivity was performed using light microscopy (Olympus, Tokyo, Japan). Two hundred cells were counted per sample to determine the percentage of positive cells (UDS detection). MMS (8 × 10-5 mol L-1; 1 h) was used as a positive control.
Cytokinesis-block micronucleus assay in V79 cells
The micronucleus assay was performed using the standard Cyt-B technique described by Matsuoka et al.36 with modifications.37 V79 cells were incubated for 4 h with various concentrations (1, 5 and 10 µmol L-1) of the test compounds. After treatment, Cyt-B was added at a final concentration of 3 µg mL-1, and the cells were harvested 21 h after Cyt-B addition. The cells were dislodged, resuspended (0.075 mol L-1 KCl at 4 ºC), and fixed with methanol/acetic acid (3:1) solution. After the slides were prepared, they were stained with Giemsa (pH 6.8). Doxorubicin (0.6 µmol L-1) was used as the positive control, and the vehicle (DMSO) was used as the negative control. Micronuclei were counted in 2,000 binucleated cells with well-preserved cytoplasm. The identification of micronuclei was performed according to the method described by Fenech.38,39
Determination of the proportion of apoptotic and necrotic cells
To observe the cell death pattern induced by the test compounds after 24 h of incubation, acridine orange/ethidium bromide (AO/EB) staining of V79 cells (3 × 105 cells mL-1) was performed according to the method described by McGahon et al.40 At least 300 cells were examined under a fluorescence microscope, and the percentages of apoptotic and necrotic cells were then calculated. Experiments were performed in triplicate in three independent experiments.
Statistical analysis
Data are presented as the mean values ± SEM (standard error of the mean). For the MTT assay, the IC50 values were obtained by nonlinear regression using the GraphPad program (Intuitive Software for Science, San Diego, CA). For other assays, data were analyzed by one-way analysis of variance (ANOVA) followed by the Newman-Keuls test.
Results and Discussion
An important strategy to obtain novel compounds with potent antitumor activities is based on the modification of β-lapachone structure.11 Among several strategies described by us, the C-ring modification (Figure 1) has been used with great success in the last years towards the synthesis of antitumor arylamino compounds applied against some cancer cell lines.11
In our preliminary studies, it was identified that some nor-β-lapachone arylamino substituted had considerably activity against HL-60 cancer cell lines. These results prompted us to evaluate the most potent compounds against one large panel of leukemia cell lines, i.e., HL-60, K562, Molt-4 and Jurkat cells. To improve our knowledge about this class of compounds, lapachones C-ring iodinated and substituted by a heterocyclic ring, 1,2,3-triazole were prepared. This pattern of substitution is based on a previous work which shows that the insertion of both 1,2,3-triazole ring and halogens in the nor-β-lapachone intensifies the desired antitumor activity.11
Table 2 shows the cytotoxic effects of nor-β-lapachone and its arylamino derivatives (1-4), methylated and iodinated naphthoquinones (5-7) and nor-β-lapachone-based 1,2,3-triazoles (8-10) on leukemia cell lines. All compounds elicited significant antiproliferative effects in all myeloid human leukemia cells examined after 72 h of exposure. Among the four leukemia cell lines evaluated, the chronic myelogenous leukemia cells (K562) were slightly less sensitive (slightly greater IC50 values in relation to the other cell lines) to treatment with the tested compounds.
The insertion of arylamino ring intensifies the activity with more efficiency than the insertion of 1,2,3-triazole ring. As observed in Table 2, the most active naphthoquinones were 1-4 with IC50 values in the range of 0.33 to 2.30 µmol L-1. The iodinated and methylated compounds 5-7 were considered less active against the evaluated cancer cell lines with IC50 values in the range of 4.05 to 3.44 µmol L-1 with some exceptions: compound 7 for HL-60 (IC50 = 1.11 µmol L-1) and compound 5 for Jurkat (IC50 = 1.38 µmol L-1).
In comparison with nor-β-lapachone, our group was delighted to see that compounds 2, 3 and 4 (for HL-60), 4 (for K562), 3 and 4 (for Molt-4) and 2, 3 and 4 (for Jurkat) were more active than nor-β-lapachone, the naphthoquinoidal precursor.
In previous studies, our research group described β-lapachone-based 1,2,3-triazoles with activity against tumor cell lines,10 and the mechanism of action is related to inducing apoptotic cell death mediated by ROS generation. In another recently study,6 our group showed a 3'-nitro-3-phenylamino nor-β-lapachone derivative with cytotoxicity based on apoptosis, which is partially caused by ROS release. In the light of previous results, the importance to verify the mechanism of action of different nor-β-lapachone arylamino substituted is relevant. Due to the potent activity observed for the compounds 1-4, these substances were selected to further studies envisaging the intrinsic mechanism of action related with the antileukemic activity observed as will be discussed in the next topics.
In general terms, when compared to 1-4, only compound 10 presented similar activities against the four cancer cell lines evaluated. This substance will be subject for further mechanism studies.
ROS are continually generated and eliminated in biological systems, playing important roles in a variety of normal biochemical functions as well as abnormal pathological processes. Excessive ROS production in the cell is known to induce apoptosis,41,42 and consequently, the ability of ROS to inflict severe cellular damage and cause cell death has been exploited as an approach to kill cancer cells. The generation of ROS associated with mitochondrial disruption, resulting in oxidative stress, has been suggested as a component of a common final pathway in the execution of the apoptotic program.43,44
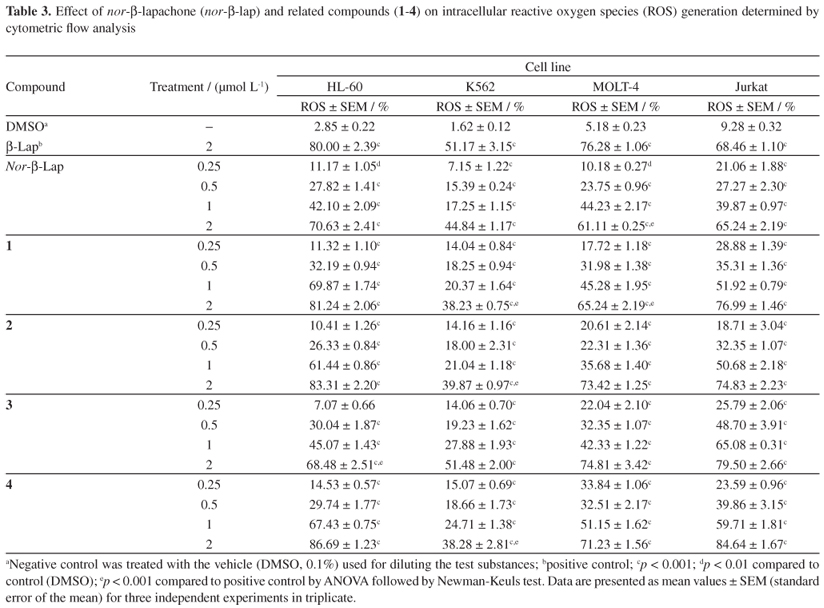
The oxidative stress associated with autooxidation of a semiquinone free radical, which produces a superoxide anion, hydrogen peroxide, and other active oxygen species, has been related to naphthoquinone cytotoxicity.45,46 Moreover, ROS have been demonstrated to be highly reactive species that cause DNA damage.47,48 In our evaluation, short exposure (4 h) to nor-β-lapachone and its derivatives (1-4) led to the generation of intracellular ROS (Table 3).
However, the levels of ROS induced after treatment were similar among all the compounds evaluated in the HL-60, MOLT-4 and Jurkat cell lines. Interestingly, the amount of ROS generated in K562 cells was lower when compared to that observed in the other three leukemia cell lines, which may be the result of high levels of GSH,49 which can confer resistance to oxidative stress.
Following short exposure (4 h) to the tested compounds, slight increases in the DNA damage index were observed for the HL-60, MOLT-4 and Jurkat cells, but not for K562 cells, as compared to doxorubicin-treated cells (Table 4).
However, there was no clear linear correlation between ROS production and the induction of DNA strand breaks after a short exposure time evaluated by standard comet assay procedure. One of the limitations of the standard comet assay, when not assaying with specific enzymes to detect oxidative damage (i.e., FPG and ENDO III), is the difficulty associated with detecting very low levels of DNA damage. At such low levels of damage, the fragments and breaks would be of such a size that migration in an agarose gel becomes difficult to visualize.50 Therefore, it was performed the modified comet assay in the presence of the endonucleases FPG and ENDOIII, which specifically recognize oxidized purine and pyrimidine bases in DNA, respectively. In our experiments, cultures treated with DMSO (0.1%) or buffer solution does not increase the DNA migration pattern in modified comet assay, in agreement with our previously publication.51Figure 2 shows the mean DNA damage caused by the tested compounds in the leukemia HL-60 and K562 cell lines, and these values are expressed as the DNA damage index after treatment with DNA repair endonucleases. Furthermore, the post-incubation results after enzyme treatment clearly showed increased DNA migration for the treated human leukemia cell cultures. The percentages of ROS generation (Table 3) in HL-60 and K562 cultures after treatments with tested compounds (2 µmol L-1) were compared with the amounts of oxidized nucleotide bases (Figure 2). In both cultures, ROS correlated with DNA damage after treatment with DNA repair enzymes (FPG and ENDOIII). Interestingly, as measured by MTT assay (Figure 3a), HL-60 cultures pre-treated with GSH-OEt showed an increased viability (close to vehicle group), which may indicate a neutralization of ROS produced by quinonoid exposure (Figure 3b), and consequently a reduction on the levels of oxidized nucleotide bases (Figure 3c), suggesting that ROS have an important role in the cytotoxic effects of tested compounds.
Some naphthoquinones have exhibited topoisomerase inhibition effects.52-54 Among these naphthoquinones, β-lapachone was first reported as a topoisomerase I catalytic inhibitor,55 and subsequently, Frydman et al.52 reported β-lapachone to be a weak topoisomerase II poison that, unlike prototypical poisons, also inactivates the enzyme upon drug/enzyme pre-incubation. However, the effects of β-lapachone on topoisomerases remain controversial, as this naphthoquinone has been shown to be both a topoisomerase I inhibitor55 and activator56 as well as a topoisomerase II inhibitor in in vitro systems.52,57 Furthermore, this agent has also been shown to fail to inhibit topoisomerase II55,56 in in vitro conditions, although this finding remains controversial.
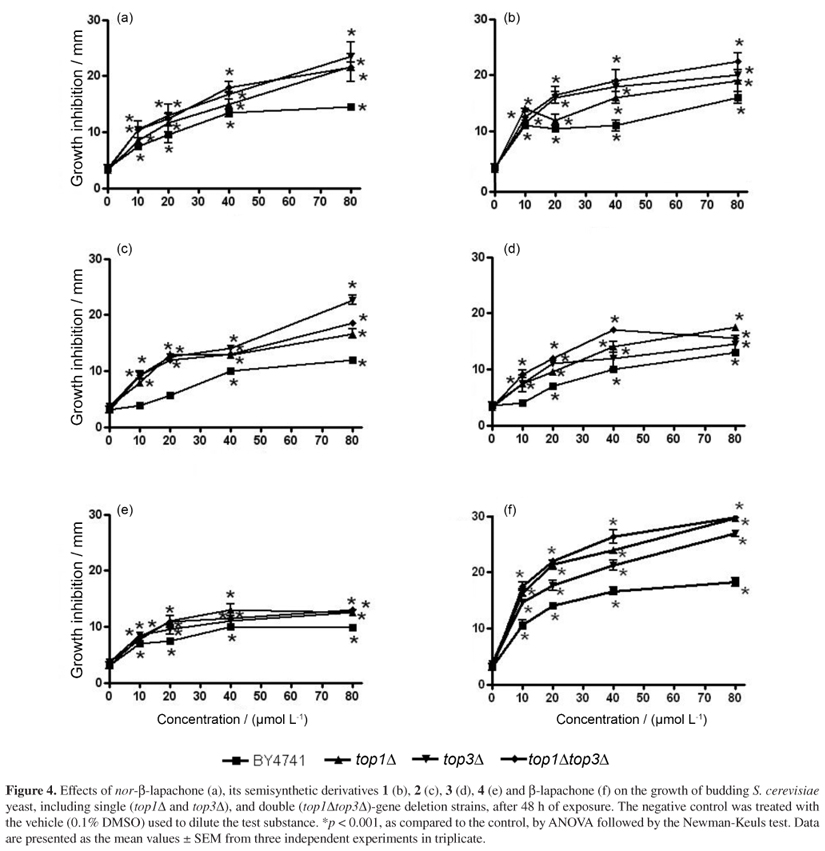
To investigate the action of nor-β-lapachone and its derivatives 1-4 on topoisomerases, it was used the budding yeast S. cerevisiae, which is commonly used to investigate the cytotoxic mechanisms of drugs that target DNA topoisomerases.58,59 Contrary to what was expected, Figure 4 demonstrates that cultures of S. cerevisiae strains with defective expression of DNA topoisomerases (top1Δ, top3Δand top1Δtop3Δ) were more sensitive to nor-β-lapachone and its derivatives (1-4) than the wild-type strain (BY4741). This result suggests that the mechanism of cytotoxicity of the naphthoquinones is, apparently, not likely to involve the inhibition of topoisomerases. The fact that the observed decrease in survival was not specific to the lack of a single type of topoisomerase can be explained by the fact that topoisomerase complexes at a DNA lesion may alter the repair efficiency and initiate alternative and more efficient processing of damage repair.60,61 Moreover, the deletion of TOP1 has no effect on yeast survival,62 furthermore, it is possible to conclude that the toxic mechanism of nor-β-lapachone and its derivatives does not include topoisomerase I poisoning. Also, our results indicate that topoisomerases served as important survival factors following DNA damage induced by the tested compounds.
It has been demonstrated that the deletion of TOP3 increased yeast sensitivity to ultraviolet light (UV)-induced DNA adducts and alkylation damage by MMS,62,63 whereas the deletion of TOP1 had no effect on yeast survival after exposure.62 This finding suggests that TOP3 may facilitate DNA damage repair, while TOP1 may hinder damage recognition.60 In addition, unlike mammalian cells, which require both type I and II topoisomerases, only the deletion of TOP2 is lethal in yeast.64 Therefore, to evaluate the action of the tested compounds on topoisomerase II, a double mutant with a deletion of TOP1 (top1Δ) and TOP3 (top3Δ) was used. The sensitivity increase of double mutant top1Δtop3Δcan be suggested as a possible role of nor-β-lapachone and their derivatives on topoisomerase II, however more studies are necessary to confirm this mechanism of action. In this sense, Krishnan and Bastow57 showed that β-lapachone was an irreversible inhibitor of topoisomerase II by induced religation and dissociation of the enzyme from DNA in the presence of ATP. Interestingly, Cline and Hanawalt60 previously showed that, similar to TOP1, TOP2 may obscure DNA lesions induced by UV damage, leading to their processing by other repair mechanisms or to the initiation of cell death if damage levels exceed the repair capabilities.
However, the growth of yeast lacking topoisomerase I expression is suppressed by β-lapachone, which raises doubts about its mechanism of action and suggests that this compound has critical intracellular targets other than topoisomerase I.57 These observations are in agreement with our data (Figure 4f) and also indicate that targets other than topoisomerase II may be associated with the cytotoxic mechanisms of β-lapachone, nor-β-lapachone and its derivatives 1-4.
Subsequent studies described β-lapachone as a DNA repair inhibitor56,65 capable of sensitizing tumor cells to DNA damaging agents.66,67β-lapachone was shown to indirectly inhibit DNA repair enzymes through the modification of the redox homeostasis of the intracellular environment, causing oxidative damage to the cysteine residues of the enzymes and subsequently impairing their structure and function to render them incapable of mediating DNA damage repair.68,69 Additionally, Neder et al.70 and Oliveira-Brett et al.71 reported that the cytotoxic actions of β-lapachone and related naphthoquinones derive, in part, from the alkylation of exposed thiol or cysteine residues on topoisomerase II. Ortho-quinones such as β-lapachone have better redox cycling ability than para-quinones such as α-lapachone. Therefore, it was suggested that the cytotoxic actions of naphthoquinones derive, at least in part, from the alkylation of exposed thiol residues on topoisomerase II-DNA complexes.70
Because β-lapachone was found to inactivate DNA repair56,65,69 and because the chemical structures of the tested compounds and β-lapachone are very similar, the potential influence of the studied naphthoquinones on the functionality of DNA repair mechanisms in HL-60 cells was tested by introducing MMS-induced DNA lesions prior to exposure to the tested compounds. MMS is an alkylating agent that directly alkylates the nitrogen of DNA bases. The repair of N-methylated base adducts in living cells occurs mainly via base excision repair, homologous recombination, and to a lesser extent, via NER.72-74
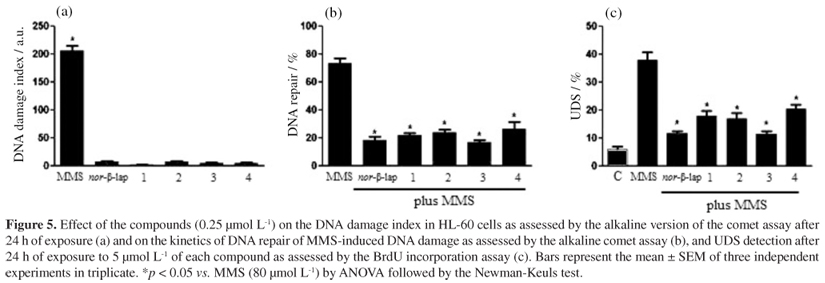
In our study, 0.25 µmol L-1 naphthoquinone did not significantly increase the DNA migration in the alkaline comet assay (Figure 5a), although it was capable of inhibiting the repair of MMS-induced DNA damage, as evidenced by the low levels of DNA repair activity observed in MMS-treated cells after post-treatment with the evaluated molecules in comparison to MMS-treated and post-incubated cells (Figure 5b). Additionally, as shown in Figure 5c, HL-60 cells treated with the tested naphthoquinones showed significantly less BrdU incorporation as compared to cells treated with MMS. These data indicated that interference with mechanisms of DNA repair (i.e., NER) might be involved in the mechanism of cytotoxic activity of these molecules.
Induction of apoptosis by naphthoquinones has been previously reported.50,75,76 Because many chemotherapeutic drugs have been shown to induce apoptosis in malignant cells, the induction of apoptosis is a target for the development of antitumor drugs.77,78 As none of the tested compounds caused nonselective growth inhibition in the leukemia cell lines evaluated, the underlying molecular mechanisms of the compound antiproliferative activity were evaluated using human promyelocytic leukemia HL-60 cells.
Our flow cytometry results with HL-60 cells showed that after pulse treatment (4 h of exposure followed by a 20 h reincubation period without the drug) of 0.5 µmol L-1 for all compounds evaluated, apoptotic events such as the alteration of cell membrane integrity (instability), mitochondrial depolarization, internucleosomal DNA fragmentation (Figure 6) and PS cell membrane localization (Figure 7) were observed (p < 0.001).
Following drug exposure, all evaluated compounds were found to slightly reduce cell membrane integrity. In addition, at least 15.54 ± 1.82% (nor-β-lapachone), 14.51 ± 0.52% (1), 15.44 ± 0.96% (2), 15.18 ± 0.44% (3), 14.75 ± 0.43% (4) and 21.18 ± 0.75% (β-lapachone) of the HL-60 cells displayed subdiploid sized DNA (sub-Go/G1), representing apoptotic cells with a fractional DNA content. The percentages of HL-60 cells showing low ΔΨm were 12.75 ± 1.12% (nor-β-lapachone), 10.84 ± 1.03% (1), 25.35 ± 1.95% (2), 12.26 ± 1.10% (3), 13.58 ± 0.76% (4), and 16.82 ± 0.81% (β-lapachone). In addition, 6.62 ± 1.19% (nor-β-lapachone), 5.79 ± 0.62% (1), 5.70 ± 0.13% (2), 2.87 ± 0.28% (3), 5.68 ± 0.64% (4) and 5.57 ± 0.37 % (β-lapachone) of the HL-60 cells demonstrated annexin V-conjugated PS in cultures treated with the lowest concentration of the test compounds. In cultures exposed to the highest concentration (2 µmol L-1) of the test substances, the percentages of HL-60 cells with degraded DNA, altered ΔΨm, and externalized PS were greater than 33, 29 and 9%, respectively, for all compounds evaluated (p < 0.001). Moreover, our data showed that after pulse treatment, lipid peroxidation (Figure 8) and nitrite/nitrate production (Figure 9) as a result of nitric oxide release were observed in treated HL-60 cells, which is in agreement with the finding that nor-β-lapachone and its arylamino derivatives (1-4) induced oxidative stress, as assessed by the determination of intracellular ROS generation detected by the H2DCFDA probe.
In situ generated ROS can open permeability transition pores with subsequent ΔΨm loss and can cause cellular damage through apoptosis, enzyme inactivation and cytochrome c release into the cytosol.79,80 In the present study, it was demonstrated that nor-β-lapachone and its arylamino derivatives (1-4) induced alterations in mitochondrial redox functions that are critical to initiating ROS-mediated cell death, in general agreement with the previously reported antiproliferative properties of other naphthoquinones.81,82 The decrease in ΔΨm induced by the test compounds indicated ROS-mediated mitochondrial dysfunction in the promyelocytic leukemia cells. Thus, the alterations in mitochondrial function caused by exposure to these naphthoquinones may be a major cause of apoptosis and cell death.
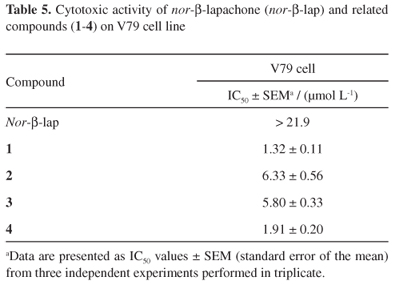
Overall, the in vivo use of quinones presents a major challenge because of their toxicity.5,83 With regard to the side effects of chemotherapy, it is essential to know whether a compound exerts a harmful effect on normal dividing cells. In this context, with the exception of nor-β-lapachone (IC50 for V79 cells was greater than 21.9 µmol L-1), all nor-β-lapachone derivatives tested exerted a cytotoxic effect against V79 cells, with the antiproliferative potential ranging from moderate (2 and 3) to high (1 and 4) (Table 5). Additionally, unlike nor-β-lapachone, a previous study determined that all four 3-arylamino-nor-β-lapachone derivatives studied here were not selective for tumor cells, as a toxic effect against proliferating human lymphocytes was observed at similar concentrations, with IC50 values after 72 h exposure equal to 2.45 ± 0.10 µmol L-1 (1), 2.49 ± 0.23 µmol L-1 (2), 3.06 ± 0.56 µmol L-1 (3) and 2.11 ± 0.21 µmol L-1 (4).
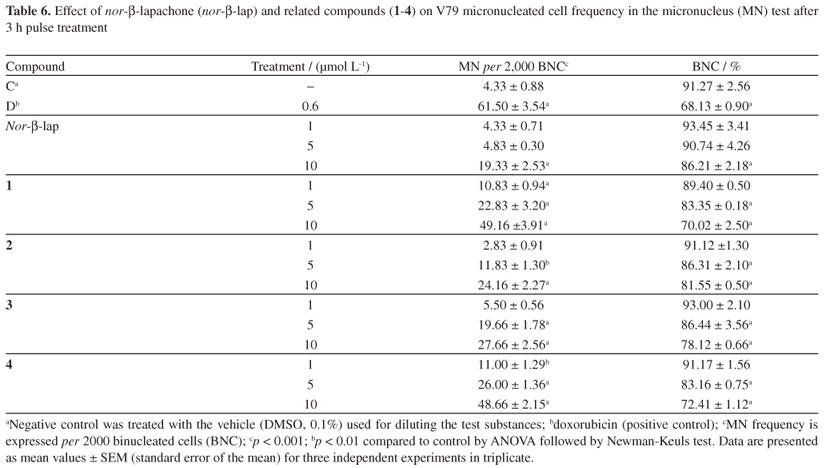
Finally, to obtain a complete evaluation of the toxic effects of these compounds in mammalian cells (V79 line), DNA damage induced by nor-β-lapachone and its arylamino derivatives (1-4) was estimated using the cytokinesis-block micronucleus test. This assay is a non-specific genotoxicity test that detects fixed mutations and permanent DNA damage. This test detects chromosomal aberrations as micronuclei in dividing versus non-dividing cells,37 which serve as a good biomarker of exposure to genotoxins. The presence of a micronucleus represents the disruption of small amounts of non-condensed chromatin, and their increased frequency is evidence of prior structural chromosome damage or changes in chromosome number.38,48 The data presented in Table 6 show that the mutagenic effects of compounds 1 and 4 occurred at even the lowest concentration tested (p < 0.001), and the effects of compounds 2 and 3 were observed at a concentration as low as 5 µmol L-1 (p < 0.001; p < 0.01, respectively), as detected by an increase in micronuclei. However, for nor-β-lapachone, increases in micronuclei were only observed at the highest concentration evaluated (p < 0.001). The reduced cytokinesis-block proliferation index (Table 6) and the presence of morphologically apoptotic nuclei in V79 cells (Figure 10) suggest that DNA damage induced by nor-β-lapachone and its arylamino derivatives (1-4) may be important features of their cytotoxic mechanisms.
Conclusion
In conclusion, the 3-arylamino-nor-β-lapachone derivatives (1-4) elicited a significant antiproliferative effect in all human myeloid leukemia cell lines examined, which is similar to the effect observed with the precursor molecule (nor-β-lapachone). Compounds 5-7 and nor-β-lapachone-based 1,2,3-triazoles 8-10 presented activity against the cancer cell lines assayed but with less activity than β-lapachone and substances 1-4. Furthermore, compounds 1-4 induce oxidative DNA damage by ROS generation, and somehow, treatments impair DNA repair activity triggering apoptosis. We hypothesized that DNA damage caused by redox cycling as a result of treatment with the tested naphthoquinones would be an important initiating signal for cell death. However, because of the lack of selectivity between cancer and non-cancer cells, the 3-arylamino-nor-β-lapachone derivatives (1-4) evaluated may have limited therapeutic potential as anticancer agents if administered as single drug but may be useful in combination with other antitumor agents. One strategy for overcoming the intrinsic toxicity of quinones may be to use derivatives that are more stable in their reduced state and thereby less likely to initiate the formation of radicals and damage cells indiscriminately. Therefore, these mechanism insights also reinforce the need for further studies with other nor-β-lapachone derivatives to determine the structure-activity relationship between both therapeutic and toxicological activities. In general terms, this work is an important contribution for developing novel antitumor drugs from lapachone group, substance currently in multiple phase II clinical trials as monotherapy and in combination with other cytotoxic drugs.84
Acknowledgments
We wish to thank Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Instituto Claude Bernard for financial support in the form of grants and fellowship awards. We are also grateful to the National Cancer Institute (Bethesda, MD, USA) for the donation of the leukemia cell lines used in this study. Dr. E. N. da Silva Júnior thanks the Programa Institucional de Auxílio à Pesquisa de Doutores Recém-Contratados and Universidade Federal de Minas Gerais (edital 12/2011), FAPEMIG (APQ-04166-10) and CNPq (Universal Project, MCTI/CNPq No. 14/2012). We also thank Silvana F. dos Santos for technical assistance and Dr. A. Leyva for English editing of the manuscript. This study is dedicated to Professor Antônio V. Pinto (in memoriam) and Maria do Carmo F. R. Pinto due to excellent advices, samples and spectra of compounds 5, 6 and 7.
Supplementary Information
Supplementary information (1H and 13C NMR spectra for all compounds) is available free of charge at http://jbcs.sbq.org.br as PDF file.
Submitted: November 5, 2012
Published online: February 19, 2013
Supplementary Information
The supplementary material is available in pdf: [Supplementary material]
- 1. Zhang, G.; Fang, L.; Zhu, L.; Zhong, Y.; Wang, P. G.; Sun, D.; J. Med. Chem. 2006, 9, 1792.
- 2. da Silva Júnior, E. N.; de Souza, M. C. B. V.; Pinto, A. V.; Pinto, M. C. F. R.; Goulart, M. O. F.; Barros, F. W.; Pessoa, C.; Costa-Lotufo, L. V.; Montenegro, R. C.; de Moraes, M. O.; Ferreira V. F.; Bioorg. Med. Chem. 2007, 15, 7035.
- 3. Joseph, P.; Long II, D. J.; Klein-Szanto, A. J. P.; Jaiswal, A. K.; Biochem. Pharmacol. 2000, 60, 207.
- 4. Wardman, P.; Curr. Med. Chem. 2001, 8, 739.
- 5. Verma, R. P.; Anticancer Agents Med. Chem. 2006, 6, 489.
- 6. Araújo, A. J.; de Souza, A. A.; da Silva Júnior, E. N.; Marinho-Filho, J. D. B.; de Moura, M. A. B. F.; Rocha, D. D.; Vasconcellos, M. C.; Costa, C. O.; Pessoa, C.; de Moraes, M. O.; Ferreira, V. F.; de Abreu, F. C.; Pinto, A. V.; Montenegro, R. C.; Costa-Lotufo, L. V.; Goulart, M. O. F.; Toxicol. in Vitro 2012, 26, 585.
- 7. Ferreira, D. C. M.; Goulart, M. O. F.; Moreira, M. S. A.; Tapsoba, I.; Arbault, S.; Bouret, Y.; Pinto, A. V.; Amatore, C.; ChemBioChem. 2009, 10, 528.
- 8. da Silva Júnior, E. N.; de Moura, M. A. B. F.; Pinto, A. V.; Pinto, M. C. F. R.; de Souza, M. C. B. V.; Araújo, A. J.; Pessoa, C.; Costa-Lotufo, L. V.; Montenegro, R. C.; de Moraes, M. O.; Ferreira, V. F.; Goulart, M. O. F.; J. Braz. Chem. Soc. 2009, 20, 635.
- 9. Viegas-Jr., C.; Danuello, A.; Bolzani, V. S.; Barreto, E. J.; Fraga, C. A.; Curr. Med. Chem. 2007, 14, 1829.
- 10. da Silva Júnior, E. N.; Cavalcanti, B. C.; Guimarães, T. T.; Pinto, M. C. F. R.; Cabral, I. O.; Pessoa, C.; Costa-Lotufo, L. V.; de Moraes, M. O.; de Andrade, C. K. Z.; dos Santos, M. R.; de Simone, C. A.; Goulart, M. O. F.; Pinto, A. V.; Eur. J. Med. Chem. 2011, 46, 399.
- 11. da Silva Júnior, E. N.; de Deus, C. F.; Cavalcanti, B. C.; Pessoa, C.; Costa-Lotufo, L. V.; Montenegro, R. C.; de Moraes, M. O.; Pinto, M. C. F. R.; de Simone, C. A.; Ferreira, V. F.; Goulart, M. O. F.; Andrade, C. K. Z.; Pinto, A. V.; J. Med. Chem. 2010, 53, 504.
- 12. da Silva Júnior, E. N.; de Melo, I. M. M.; Diogo, E. B. T.; Costa, V. A.; de Souza Filho, J. D.; Valença, W. O.; Camara, C. A.; de Oliveira, R. N.; de Araújo, A. S.; Emery, F. S.; dos Santos, M. R.; de Simone, C. A.; Menna-Barreto, R. F. S.; de Castro, S. L.; Eur. J. Med. Chem. 2012, 52, 304.
- 13. Silva, R. S. F.; Costa, E. M.; Trindade, U. L. T.; Teixeira, D. V.; Pinto, M. C. F. R.; Santos, G. L.; Malta, V. R. S.; de Simone, C. A.; Pinto, A. V.; Castro, S. L.; Eur. J. Med. Chem. 2006, 41, 526.
- 14. Fieser, L. F.; Fieser, M.; J. Am. Chem. Soc. 1948, 70, 3215.
- 15. Rostovtsev, V. V.; Green, G. L.; Fokin, V. V.; Sharpless, K. B.; Angew. Chem., Int. Ed. 2002, 41, 2596.
- 16. Mosmann, T.; J. Immunol. Methods 1983, 16, 55.
- 17. Crow, J. P.; Nitric Oxide 1997, 1, 145.
- 18. Hempel, S. L.; Buettner, G. R.; O'Malley, Y. Q.; Wessels, D. A.; Flaherty, D. M.; Free Radical Biol. Med. 1999, 27, 146.
- 19. LeBel, C. P.; Ischiopoulos, H.; Bondy, S. C.; Chem. Res. Toxicol. 1992, 5, 227.
- 20. Singh, N. P.; McCoy, M. T.; Tice, R. R.; Schneider, E. L.; Exp. Cell Res. 1988, 175, 184.
- 21. Klaude, M.; Eriksson, S.; Nygren, J.; Ahnström, G.; Mutat. Res. 1996, 363, 89.
- 22. Tice, R. R., Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J. C.; Sasaki, Y. F.; Environ. Mol. Mutagen. 2000, 35, 206.
- 23. Miyamae, Y.; Yamamoto, M.; Sasaki, Y. F.; Kobayashi, H.; Igarashi-Soga, M.; Shimoi, K.; Hayashi, M.; Mutat. Res. 1998, 418, 131.
- 24. Hartmann, A.; Agurell, E.; Beevers, C.; Brendler-Schwaab, S.; Burlinson, B.; Clay, P.; Collins, A.; Smith, A.; Speit, G.; Thybaud, V.; Tice, R. R.; Mutagenesis 2003, 18, 45.
- 25. Cavalcanti, B. C.; Bezerra, D. P.; Magalhães, H. I. F.; de Moraes, M. O.; Lima, M. A.; Silveira, E. R.; Câmara, C. A.; Rao, V. S.; Pessoa, C.; Costa-Lotufo, L. V.; J. Appl. Toxicol. 2009, 29, 560.
- 26. Gietz, R. D.; Woods, R. A.; Methods Enzymol. 2002, 350, 87.
- 27. Collins, S. J.; Blood 1987, 70, 1233.
- 28. Militão, G. C. G.; Dantas, I. N. F.; Pessoa, C.; Falcão, M. J.; Silveira, E. R.; Lima, M. A.; Curi, R.; Lima, T.; de Moraes, M. O.; Costa-Lotufo, L. V.; Life Sci. 2006, 78, 2409.
- 29. Draper, H. H.; Hadely, M.; Methods Enzymol. 1990, 186, 421.
- 30. Green, L. C.; Tannenbaum, S. R.; Goldman, P.; Science 1981, 212, 56.
- 31. Logrado, L. P.; Santos, C. O.; Romeiro, L. A.; Costa, A. M.; Ferreira J. R.; Cavalcanti, B. C.; de Moraes, O. M.; Costa-Lotufo, L. V.; Pessoa, C.; dos Santos, M. L.; Eur. J. Med. Chem. 2010, 45, 3480.
- 32. Marinho-Filho, J. D.; Bezerra, D. P.; Araújo, A. J.; Montenegro, R. C.; Pessoa, C.; Diniz, J. C.; Viana, F. A.; Pessoa, O. D.; Silveira, E. R.; de Moraes, M. O.; Costa-Lotufo, L. V.; Chem. Biol. Interact. 2010, 183, 369.
- 33. Cavalcanti, B. C.; da Costa, P. M.; Carvalho, A. A.; Rodrigues, F. A.; Amorim, R. C.; Silva, E. C.; Pohlit, A. M.; Costa-Lotufo, L. V.; de Moraes, M. O.; Pessoa, C.; Pharm. Biol. 2012, 50, 980.
- 34. Bootsma, D.; Mudle, M. P.; Cohen, J. A.; Pot F.; Mutat. Res. 1970, 9, 507.
- 35. Snyder, R. D.; Mutat. Res. 1984, 131, 163.
- 36. Matsuoka, A.; Yamazaki, N.; Suzuki, T.; Hayashi, M.; Sofuni, T.; Mutat. Res. 1992, 272, 223.
- 37. Bonacker, D.; Stoiber, T., Wang, M.; Böhm, K. J.; Prots, I.; Unger, E.; Their, R.; Bolt, H. M.; Degen, G. H.; Arch. Toxicol. 2004, 78, 575.
- 38. Fenech, M.; Mutat. Res. 2000, 455, 81.
- 39. Fenech, M.; Mutat. Res. 2006, 600, 58.
- 40. McGahon, A. J.; Martin, S. M.; Bissonnette, R. P.; Mahboubi, A.; Shi, Y.; Mogil, R. J.; Nishioka, W. K.; Green, D. R.; Methods Cell Biol. 1995, 46, 153.
- 41. Sastre, J.; Pallardó, F. V.; Viña, J.; IUBMB Life 2000, 49, 427.
- 42. Martindale, J. L.; Holbrook, N. J.; J. Cell Physiol. 2002, 192, 1.
- 43. Buttke, T. M.; Sandstrom, P. A.; Immunol. Today 1994, 15, 7.
- 44. Slater, A. F.; Stefan, C.; Nobel, I.; Van den Dobbelsteen, D. J.; Orrenius, S.; Toxicol. Lett. 1995, 82-83, 149.
- 45. Cadenas, E.; Biochem. Pharmacol. 1995, 49, 127.
- 46. Fourie, J.; Oleschuk, C. J.; Guziec, F.; Guziec, L.; Derek, J.; Fiterman, D. J.; Monterrosa, C.; Begleiter, A.; Cancer Chemother. Pharmacol. 2002, 49, 101.
- 47. Devasagayam, T. P. A.; Steenken, S.; Obendorf, M. S. W.; Schulz, W. A.; Sies, H.; Biochemistry 1991, 30, 6283.
- 48. Cavalcanti, B. C.; Nobre Jr., H. V.; Seleghim, M. H. R.; Berlinck, R. G.; Cunha, G. M.; de Moraes, M. O.; Pessoa, C.; Chem.-Biol. Interact. 2008, 174, 155.
- 49. Chau, Y. P.; Shiah, S. G.; Don, M. J.; Kuo, M. L.; Free Radical Biol. Med. 1998, 24, 660.
- 50. Olive, P. L.; Durand, R. E.; Cytometry A 2005, 66, 1.
- 51. Cavalcanti, B. C.; Barros, F. W. A.; Cabral, I. O.; Ferreira, J. R. O.; Magalhães, H. I. F.; Nobre Júnior, H. V. N.; da Silva Júnior, E. N.; de Abreu, F. C.; Costa, C. O.; Goulart, M. O. F.; Moares, M. O.; Pessoa, C.; Chem. Res. Toxicol. 2011, 24, 1560.
- 52. Frydman, B.; Marton, L. J.; Sun, J. S.; Neder, K.; Witiak, D. T.; Liu, A. A.; Wang, H.; Mao, Y.; Wu, H.; Sanders, M. M.; Liu, L. F.; Cancer Res. 1997, 57, 620.
- 53. Krishnan, P.; Bastow, K. F.; Cancer Chemother. Pharmacol. 2001, 47, 187.
- 54. Sperry, J.; Lorenzo-Castrillejo, I.; Brimble, M. A.; Machín, F.; Bioorg. Med. Chem. 2009, 17, 7131.
- 55. Li, C. J.; Averboukh, L.; Pardee, A. B.; J. Biol. Chem. 1993, 268, 22463.
- 56. Boothman, D. A.; Trask, D. K.; Pardee, A. B.; Cancer Res. 1989, 49, 605.
- 57. Krishnan, P.; Bastow, K. F.; Biochem. Pharmacol. 2000, 60, 1367.
- 58. Reid, R. J. D.; Benedetti, P.; Bjornsti, M. A.; Biochim. Biophys. Acta 1998, 1400, 289.
- 59. Rogojina, A. T.; Nitiss, J. L.; J. Biol. Chem. 2008, 283, 29239.
- 60. Cline, S. D.; Hanawalt, P. C.; DNA Repair 2006, 5, 611.
- 61. Wagner, M.; Price, G.; Rothstein, R.; Genetics 2006, 174, 555.
- 62. Nitiss, J. L.; Nitiss, K. C.; Rose, A.; Waltman, J. L.; J. Biol. Chem. 2001, 276, 26708.
- 63. Oakley, T. J.; Hickson, I. D.; DNA Repair 2002, 1, 175.
- 64. Nitiss, J. L.; Biochim. Biophys. Acta 1998, 1400, 63.
- 65. Boothman, D. A.; Schlegel, R.; Pardee, A. B.; Mutat. Res. 1988, 202, 393.
- 66. Boothman, D. A.; Greer, S.; Pardee, A. B.; Cancer Res. 1987, 47, 5361.
- 67. Li, C. J.; Wang, C.; Pardee, A. B.; Cancer Res. 1995, 55, 3712.
- 68. Boorstein R. J.; Pardee, A. B.; Biochem. Biophys. Res. Commun. 1984, 118, 828.
- 69. Bentle, M. S.; Reinicke, K. E.; Bey, E. A.; Spitz, D. R.; Boothman, D. A.; J. Biol. Chem. 2006, 281, 33684.
- 70. Neder, K.; Marton, L. J.; Liu, L. F.; Frydman, B.; Cell Mol. Biol. 1998, 44, 465.
- 71. Oliveira-Brett, A. M.; Goulart, M. O. F.; Abreu, F. C.; Bioelectrochemistry 2002, 56, 53.
- 72. Sancar, A.; Lindsey-Boltz, L. A.; Unsal-Kacmaz, K.; Linn, S.; Annu. Rev. Biochem. 2004, 73, 39.
- 73. Meira, L. B.; Burgis, N. E.; Samson, L. D.; Adv. Exp. Med. Biol. 2005, 570, 125.
- 74. Schwartz, T. R.; Kmiec, E. B.; Gene Ther. Mol. Biol. 2005, 9, 193.
- 75. Kumar, M. R. S.; Aithal, K.; Rao, B. N.; Udupa, N.; Rao, B. S.; Toxicol. In Vitro 2009, 23, 242.
- 76. D'anneo, A.; Augello, G.; Santulli, A.; Giuliano, M.; Di Fiore, R.; Messina, C.; Tesoriere, G.; Vento, R.; J. Cell Physiol. 2010, 222, 433.
- 77. Bamford, M.; Walkinshaw, G.; Brown, R.; Exp. Cell Res. 2000, 256, 1.
- 78. Fernandez-Luna, J. L.; Clin. Transl. Oncol. 2007, 9, 555.
- 79. Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S. M.; Ahmad, M.; Alnemri, E. S.; Wang, X.; Cell 1997, 91, 479.
- 80. Pisani, D. E.; Elliot, A. J.; Hinman, D. R.; Aaronson, L. M.; Pardini, R. S.; Biochem. Pharmacol. 1986, 35, 3791.
- 81. Yu-Bin, J.; Zhong-Yuan, Q.; Xiang, Z.; Exp. Toxicol. Pathol. 2011, 63, 69.
- 82. Bolton, J. L.; Trush, M. A.; Penning, T. M.; Dryhurst, G.; Monks, T. J.; Chem. Res. Toxicol. 2000, 13, 135.
- 83. Miao, X. S.; Zhong, C.; Wang, Y.; Savage, R. E.; Yang, R. Y.; Kizer, D.; Volckova, E.; Ashwell, M. A.; Chan, T. C.; Rapid Commun. Mass Spectrom. 2009, 23, 12.
- 84. Zakharova, O. D.; Ovchinnikova, L. P.; Goryunov, L. I.; Troshkova, N. M.; Shteingarts, V. D.; Nevinsky, G. A.; Eur. J. Med. Chem. 2010, 45, 2321.
Publication Dates
-
Publication in this collection
28 Feb 2013 -
Date of issue
Jan 2013
History
-
Received
05 Nov 2012 -
Accepted
19 Feb 2013