Abstract
For the first time, the adsorption of pesticides such as azinphos-methyl and methamidophos by polyamidoamine (PAMAM) derivatives was studied. Amine groups of PAMAM (G4 and G5) were functionalized with different biomolecules such as folic acid, coumarine, arginine, lysine, and asparagine. Subsequently, the synthesized compounds were used to trap organophosphates (OP), and its affinity to do so was measured by high-performance liquid chromatography (HPLC). The obtained experimental data was compared with the interaction energy values obtained through a nanoinformatic methodology, by using conformational sampling through Euler angles and semi‑empirical quantum mechanical calculations. Both, the experimental and the in silico methodology can be employed to screen with high accuracy the molecular interactions between OP agents and the functionalized PAMAM. Furthermore, affinity results by HPLC and molecular dynamics were supported by in vitro enzyme acetylcholinesterase activity assays.
polyamidoamine (PAMAM); azinphos-methyl (AZM); methamidophos (MMP); pesticides; acetylcholinesterase activity (AChE); semi-empirical calculations
Introduction
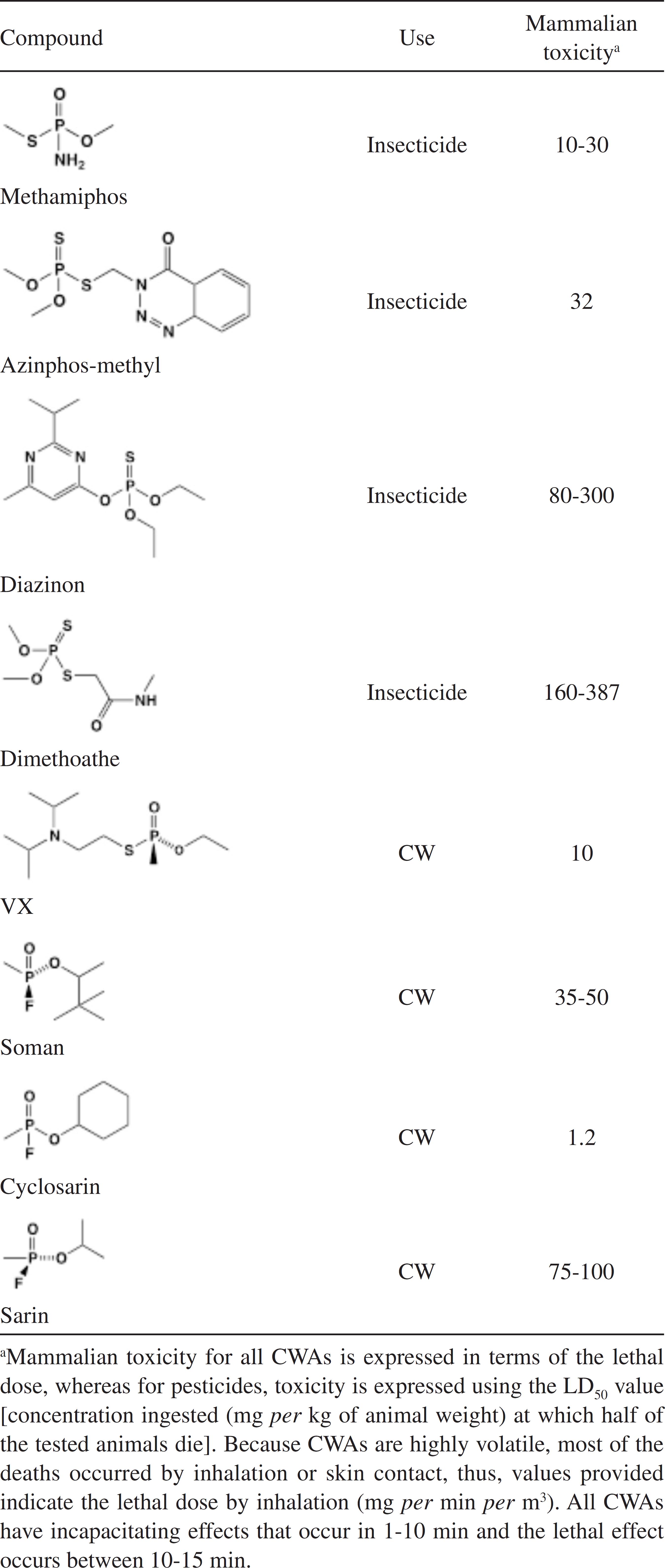
Worldwide, the most employed insecticides for indoor use and agricultural purposes belong to carbamates, pyrethroids or organophosphates (OP).11 Sogorb, M. A.; Vilanova, E.; Toxicol. Lett. 2002, 128, 215. As pesticides, OP have been used since the end of the Second World War. But OP have also been used as lethal nerve and chemical warfare agents (CWAs), such as VX, soman and sarin.22 Obare, S. O.; De, C.; Guo, W.; Haywood, T. L.; Samuels, T. A.; Adams, C. P.; Masika, N. O.; Murray, N. O. D. H.; Anderson, G. A.; Campbell, K.; Fletcher, K.; Sensors (Basel) 2010, 10, 7018. OP pesticides and CWAs have similar chemical structures (Table 1) and therefore have similar mechanisms of toxicity.33 Singh, B. K.; Nat. Rev. Microbiol. 2009, 7, 156. This is based on its capacity to bind the active site of acetylcholinesterase (AChE), resulting on an accumulation of acetylcholine at the neuron synapses.44 Emerick, G. L.; DeOliveira, G. H.; Oliveira, R. V.; Ehrich, M.; Toxicology 2012, 292, 145.,55 Saheli, P.; Indrani, R.; Gopal, L.; Prasun, P.; Samrat, R.; Arunava, S.; Arunava, G.; J. Environ. Sci. Health, Part B 2013, 48, 559. The neurotransmitter accumulation leads to symptoms related to the autonomous nervous system (abdominal cramps, nausea, diarrhea and salivation) and the central nervous system (dizziness, tremor, anxiety and confusion).66 Montesano, M. A.; Olsson, A. O.; Kuklenyik, P.; Needham, L. L.; Bradman, A.; Barr, D. B.; J. Exposure Sci. Environ. Epidemiol. 2007, 17, 321.,77 Sogorb, M. A.; Vilanova, E.; Carrera, V.; Toxicol. Lett. 2004, 151, 219. Due to this, one of the current greatest threats in modern world is the possibility of OPs use as chemical weapons by terrorist groups.88 Delfino, R. T.; Ribeiro, T. S.; Figueroa-Villar, J. D.; J. Braz. Chem. Soc. 2009, 20, 407.
The World Health Organization estimates that around 3 million people per year are poisoned by pesticides, most of them by OP pesticides, resulting in around 200,000 deaths.99 Bird, S. B.; Sutherland, T. D.; Gresham, C.; Oakeshott, J.; Scott, C.; Eddleston, M.; Toxicology 2008, 247, 88. Poisoning occurs by accidental spillage, suicide attempts and occupational hazards involving workers and farmers.33 Singh, B. K.; Nat. Rev. Microbiol. 2009, 7, 156.
Nanotechnology has become an area of great interest in nanomedicine and life science applications as drug delivery, gene delivery, or nanodiagnosis systems,1010 Esfand, R.; Tomalia, D. A.; Drug Discovery Today
2001, 6, 427.
11 Navarro, G.; de ILarduya, C. T.; Nanomed. Nanotechnol. Biol. Med. 2009, 5, 287.-1212 Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem.
2011, 400, 483. but there are few studies that use dendrimers as nano-detoxification agents of harmful compounds in the body. For this reason, OPs' capture, through the use of nanocompounds, is a growing and interesting field of development, due to its capacity for selective capture of toxic compounds in vivo. The aim of this study is to evaluate the ability of dendrimers to capture OPs in a selective manner. Polyamidoamine (PAMAM) dendrimers are a class of artificial macromolecules that have shown excellent potential in biomedical applications due to their unique physical and chemical properties. Among these properties, PAMAM show a precise control on molecular size, shape and density, high solubility, a large number of surface functional groups and hydrophobic cavities that can capture molecules of interest.1313 Dong, Z.; Katsumi, H.; Sakane, T.; Yamamoto, A.; Int. J. Pharm. 2010, 293, 244. PAMAM are biocompatible, non-immunogenic, water-soluble, and possess terminal modifiable amine functional groups for binding various targeting or guest molecules.1414 Devarakonda, B.; Hill, B. R. A.; de Villiers, M. M.; Int. J. Pharm. 2004, 284, 133. PAMAM are large, highly branched macromolecules that are synthesized from an ethylenediamine core by a successive addition of methyl acrylate and ethylenediamine. The number of terminal amino groups increase as the generation of PAMAM increases.1515 Domanski, D. M.; Klajnert, B.; Bryszewska, M.; Bioelectrochem. 2004, 63, 189.
The OPs, such as azinphos-methyl (AZM) and methamidophos (MMP), are some of the most commonly used pesticides in agriculture and domestic purposes.1616 Jaga, K.; Dharmani, C.; Rev. Panam. Salud Publica
2003, 14, 171.
17 Kamanyire, R.; Karalliedde, L.; Occup. Med-Oxford
2004, 54, 68.-1818 Lima, C. S.; Dutra-Tavares, A. C.; Nunes, F.; Nunes-Freitas, A. L.; Ribeiro-Carvalho, A.; Filgueiras, C. C.; Manhaes, A. C.; Meyer, A.; Abreu-Villaca, Y.; Toxicol. Sci. 2013, 134, 125. Its easy acquisition and handling can pose a great threat if used as CW, and the potential attack employing these agents gives a strong reason for continuous research on the development of more effective antidotes against them.
Structure-based molecular characterization using semi-empirical quantum mechanical studies1919 Stewart, J. J.; J. Mol. Model. 2007, 13, 1173.,2020 Korth, M.;J. Chem. Theory Comput. 2010, 6, 3808. can be used to accurately understand the properties that govern the interactions between molecular systems. However, the application of these computational chemistry methods to analyze nanostructured systems requires its suitable adaptation and implementation. To solve the above problem, a new discipline has emerged, called “nanoinformatics”,2121 Gonzalez-Nilo, F.; Perez-Acle, T.; Guinez-Molinos, S.; Geraldo, D. A.; Sandoval, C.; Yevenes, A.; Santos, L. S.; Laurie, V. F.; Mendoza, H.; Cachau, R. E.; Biol. Res. 2011, 44, 43. which meets all the advances in information technology, nanotechnology and bioinformatics to be applied in nanoscale research.2222 Maojo, V.; Fritts, M.; de la Iglesia, D.; Cachau, R. E.; Garcia-Remesal, M.; Mitchell, J. A.; Kulikowski, C.; Int. J. Nanomed.
2012, 7, 3867.
23 Maojo, V.; Fritts, M.; Martin-Sanchez, F.; de la Iglesia, D.; Cachau, R. E.; Garcia-Remesal, M.; Crespo, J.; Mitchell, J. A.; Anguita, A.; Baker, N.; Barreiro, J. M.; Benitez, S. E.; de la Calle, G.; Facelli, J. C.; Ghazal, P.; Geissbuhler, A.; Gonzalez‑Nilo, F.; Graf, N.; Grangeat, P.; Hermosilla, I.; Hussein, R.; Kern, J.; Koch, S.; Legre, Y.; Lopez-Alonso, V.; Lopez-Campos, G.; Milanesi, L.; Moustakis, L.; Munteanu, C.; Otero, P.; Pazos, A.; Perez-Rey, D.; Potamias, G.; Sanz, F.; Kulikowski, C.; Comput. Sci. Eng.
2012, 94, 521.-2424 de la Iglesia, D.; Maojo, V.; Chiesa, S.; Martin‑Sanchez, F.; Kern, J.; Potamias, G.; Crespo, J.; Garcia-Remesal, M.; Keuchkerian, S.; Kulikowski, C.; Mitchell, J. A.; Methods Inf. Med. 2011, 50, 84.
In this work, a nanoinformatics methodology was used to calculate the interaction energy between molecule(1)- molecule(2) pairs.2525 Fan, C. F.; Olafson, B. D.; Blanco, M.; Hsu, S. L.; Macromol. 1992, 25, 3667.,2626 Avila-Salas, F.; Sandoval, C.; Caballero, J.; Guinez‑Molinos, S.; Santos, L. S.; Cachau, R. E.; Gonzalez-Nilo, F. D.; J. Phys. Chem. B 2012, 116, 2031. The first molecule represents the monomeric unit of a specific dendrimer attached to a functional group, and the second molecule represents the OP compound. To obtain an accurate prediction of the interaction energies, the nanoinformatics methodology implements a Metropolis Monte Carlo algorithm2727 Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.; Teller, E.; J. Chem. Phys. 1953, 21, 1087. that performs a random sampling through Euler angles2525 Fan, C. F.; Olafson, B. D.; Blanco, M.; Hsu, S. L.; Macromol. 1992, 25, 3667. to generate thousands of different molecular conformations (for a specific pair), then for each new conformation, its interaction energy is calculated using a semi-empirical quantum mechanical method.1919 Stewart, J. J.; J. Mol. Model. 2007, 13, 1173.,2020 Korth, M.;J. Chem. Theory Comput. 2010, 6, 3808. The long list of interaction energy values can be used for in silico analysis of the affinity between OP agents and the functionalized PAMAM. Thus, in silico techniques are perfect tools for this task since they can quickly provide structures of several new lethal compounds for further experimental work.2828 França, T. C.; Guimarães, A. P.; Cortopassi, W. A.; Oliveira, A. A.; Ramalho, T. C.; Curr. Comput.-Aided Drug Des. 2013, 9, 507.
The aim of the present work is to incorporate active biomolecules such as folic acid, coumarin, lysine, arginine and asparagine as new end-groups in the structures of PAMAM generation 4 and 5 (G4 and G5) to increase the capacity to capture highly toxics OP compounds, specifically, AZM and MMP.
Methodology
Materials
N-hydroxybenzotriazole (HOBt), 1-[3-(dimethylamino) propyl]-3-ethylcarbodi-imide hydrochloride (EDC·HCl), folic acid (FA), Boc-Arginine(Tos)-OH [Boc‑Arg(Tos)- OH], Boc-Lysine(Z)-OH [Boc-Lys(Z)-OH], Boc‑Asparagine(Trt)‑OH [Boc-Asn(Trt)-OH], coumarin- 3-carboxylic acid (Cou), dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF) were purchased from Sigma Aldrich Co. (Saint-Louis, MO, USA). Dialysis membrane (M.W. cut-off of 500 Da) was purchased from Spectrum Labs ADP. Methanol (HPLC grade) was purchased from Merck (Germany). Dendrimers PAMAM G4 and G5 were purchased from Dendritech, Inc. (USA). Pure azinphos-methyl-[O,O-dimethyl-S-(3,4-dihydro-4- keto-1,2,3-benzotriazinyl-3-methyl) dithiophosphate] and methamidophos (O,S-dimethylphosphoramidothioate) were purchased Sigma-Aldrich.
MALDI analysis
To confirm the molecular weight of surface modified PAMAM, mass spectra analyses of the dendrimers were performed on matrix-assisted laser desorption ionizationtime of flight (MALDI-TOF) equipment (Bruker Autoflex) with a pulsed nitrogen laser (337 nm), operating in positive ion reflector mode, using 19 kV acceleration voltage and 2,5-dihydroxybenzoic acid (DHB) as matrix.
Synthesis of PAMAM derivatives
Six different PAMAM formulations varying size and surface charges were synthetized as described below.
PAMAM-G5-Folate derivative (G5-FA): conjugation of G5 with FA was carried out by a condensation between the γ-carboxyl group of FA and the primary amino group of PAMAM, according to the previously reported method1212 Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem. 2011, 400, 483.,2929 Stewart, J. J. P.; MOPAC2009, Version 10.040L; Stewart Computational Chemistry: Colorado Springs, USA, 2009. [available as Supplementary Information (SI)].
PAMAM-Coumarine derivative (G5-Cou): conjugation of G5 with Cou was carried out by a condensation between the carboxyl group of Cou and the primary amino group of PAMAM, according to the previously reported method1212 Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem. 2011, 400, 483.,3030 Pisal, D. S.; Yellepeddi, V. K.; Kumar, A.; Kaushik, R. S.; Hildreth, M. B.; Guan, X.; Palakurthi, S.; Int. J. Pharm. 2008, 350, 113. (available as SI).
PAMAM G4-Arginine(Tos)-OH (G4-Arg): conjugation of G4 with Boc-Arg(Tos)-OH was carried out by a condensation between the carboxyl group of arginine and the primary amino group of PAMAM, according to the previously reported method1212 Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem. 2011, 400, 483.,3030 Pisal, D. S.; Yellepeddi, V. K.; Kumar, A.; Kaushik, R. S.; Hildreth, M. B.; Guan, X.; Palakurthi, S.; Int. J. Pharm. 2008, 350, 113. (available as SI).
PAMAM G4-Lysine(Z)-OH (G4-Lys): conjugation of G4 with Boc-lys(Z)-OH was synthesized according to the previously reported method1212 Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem. 2011, 400, 483.,3030 Pisal, D. S.; Yellepeddi, V. K.; Kumar, A.; Kaushik, R. S.; Hildreth, M. B.; Guan, X.; Palakurthi, S.; Int. J. Pharm. 2008, 350, 113. (available as SI).
PAMAM G4-Arg(Tos)-OH/Lys(Z)-OH (PAMAM G4‑Arg/ Lys): the functionalization of G4 with Boc‑Arg(Tos)‑OH and Boc-Lys(Z)-OH, was synthesized according to the previously reported method1212 Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem. 2011, 400, 483.,3030 Pisal, D. S.; Yellepeddi, V. K.; Kumar, A.; Kaushik, R. S.; Hildreth, M. B.; Guan, X.; Palakurthi, S.; Int. J. Pharm. 2008, 350, 113. (available as SI).
Affinity assays
For each PAMAM and functionalized PAMAM, experiments were carried out to determine the extent of binding and fractional binding of MMP in methanol solutions. Briefly, functionalized PAMAM adjusted at 5.83 ˟ 10-3 mmol L-1 was mixed in a 1:1 v/v ratio with a 0.07 mmol L-1 (10 ppm) of MMP to give a final MMP/PAMAM mol L-1 ratio of 12:1. Assays were performed at pH 4.5-5.5. The samples were mixed for 45 min with the PAMAM derivatives at constant room temperature (25 °C), and then centrifuged at 10000 rpm for 10 min. The concentrations of MMP in supernatants separated from precipitated were analyzed by HPLC. The adsorption efficiency of each OP compound by PAMAM were evaluated by determining the percentage decrease in the absorbance at each specific maximum absorbance wavelength using the following equation:

where A0 is the initial absorbance at specific wavelength and A is the final absorbance at the same wavelength of each OP.3131 Durán-Lara, E. F.; López-Cortés, X. A.; Castro, R. I.; Ávila-Salas, F.; González-Nilo, F. D.; Laurie, F. V.; Santos, L. S.; Food Chem. 2015, 168, 464.
Chromatographic method
An Agilent 1260 Infinity HPLC system (USA) with a photodiode array detector and a C-18 Lichrospher 100 RP‑18 (250 mm ˟ 4 mm i.d. ˟ 5 μm) column was used for the analysis of column eluents.
A 40 μL solution of the studied supernatant was injected into the HPLC apparatus. An isocratic elution with methanol-water mixture (70:30, v/v) at a flow rate of 1.5 mL min-1 was used as mobile phase. The eluents were monitored at 230 nm by absorbance detection. For data processing, the chromatographic software ChemStation was used. The chromatographic determination was performed at room temperature. Reproducibility of the method was determined for individual standards. Three replicates per pesticide and concentration level were prepared.
Calibration procedures
AZM and MMP standards were prepared in methanol. The concentrations ranged from 10, 5.0, 2.5 and 1.25 ppm. Linear calibration curves were obtained by plotting the peak areas of the individual chemicals as a function of the concentration using the GraphPad Prism program for Windows (GraphPad Software, Inc., San Diego, CA, U.S.A.).
Statistical analysis
All the experiments were carried out in triplicate and the student t-test was used to calculate the statistical differences between the experimental compound and the negative control. A p-value of < 0.05 was considered significant.
Nanoinformatics studies
Theoretical platform
Two OP compounds were chosen for detailed molecular dynamic (MD) investigations, MMP and AZM. G4-Asn10, which presented the highest binding capacity for MMP at pH 7, was chosen for build systems and was parameterized according to the rules for all-atom CHARMM27.3232 Phillips, J. C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R. D.; Kale, L.; Schulten, K.; J. Comput. Chem. 2005, 26, 1781. This force field was not included in conventional force field due to the dendritic structures. Charges, missing bond, angle, dihedral parameters of the PAMAM core, as well as intermediate dendrons and the outer surface were estimated from similar terms within the force field. An atomistic model for G4-Asn10 was built at pH 7 containing 64 protonated amines in total (54 terminal amines from PAMAM and 10 amines from Asn end groups). The 10 units of Asn functional groups have been homogeneously distributed in 64 end groups. The molecular parameters for MMP and AZM were obtained from SwissParam3333 Zoete, V.; Cuendet, M. A.; Grosdidier, A.; Michielin, O.; J. Comput. Chem. 2001, 32, 2359. online server (http://www.swissparam.ch/) by using the Merck Molecular Force Field (MMFF) at pH 7 (net charge = 0 for both molecules). The G4-Asn10 structure and MMP and AZM molecules were implemented for two systems. The first system contained 50 molecules of MMP and 1 molecule of PAMAM (MMP: PAMAM, 50:1) and the second one contained 50 molecules of AZM and 1 molecule of PAMAM (AZM: PAMAM, 50:1) in order to evaluate the interaction of MMP and AZM molecules over the entire surface of the PAMAM. For these models, G4-Asn10 was set to the origin of the coordinate system. Subsequently, the PAMAM was solvated in a water periodic box (104 ˟ 107 ˟ 101 Å3) of TIP3 water molecules,3434 Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L.; J. Chem. Phys. 1983, 79, 926. then the corresponding number of MMP and AZM molecules were added in a random way along the water box (Figure 1). In order to neutralize the system, 64 chloride ions were added for the first and second system, respectively. Both systems were minimized for 10,000 steps and equilibrated through MD simulation. The MD simulations were performed at 298 K in the isobaric-isothermal ensemble for 20 ns. The temperature was maintained with the Langevin thermostat with a damping coefficient of 1 ps-1 whereas the pressure (1 atm) was kept constant using the Langevin piston method.3535 Feller, S.; Zhang, Y.; Pastor, R.; Brooks, B.; J. Chem. Phys. 1995, 103, 4613. Equations of motion were integrated with a 2 fs time step. The van der Waals (vdW) cut-off was set to 12 and the long-range electrostatic forces were computed using the particle-mesh Ewald approach. The MD simulation was performed using the NAMD2.93131 Durán-Lara, E. F.; López-Cortés, X. A.; Castro, R. I.; Ávila-Salas, F.; González-Nilo, F. D.; Laurie, F. V.; Santos, L. S.; Food Chem. 2015, 168, 464. program and CHARMM27 force field3636 MacKerell Jr., A. D.; Bashford, D.; Bellott, M.; Dunbrack, R. L.; Evanseck, J. D.; Field, M. J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; Joseph-McCarthy, D.; Kuchnir, L.; Kuczera, K.; Lau, F. T. K.; Mattos, C.; Michnick, S.; Ngo, T.; Nguyen, D. T.; Prodhom, B.; Reiher, W. E.; Roux, B.; Schlenkrich, M.; Smith, J. C.; Stote, R.; Straub, J.; Watanabe, M.; Wiórkiewicz-Kuczera, J.; Yin, D.; Karplus, M.; J. Phys. Chem. B 1998, 102, 3586. for standard ions and water molecules. All analyses were done with VMD3737 Humphrey, W.; Dalke, A.; Schulten, K.; J. Mol. Graphics 1996, 14, 33. software. For the analysis of the molecular dynamics, the data collected along the trajectories were used to calculate the radius of gyration, Rg, of the PAMAM and different concepts were applied such as radial pair distribution function (RDF) of MMP and AZM molecules. Furthermore, several scripts were implemented in order to get relevant information from de MD, such as capture of MMP and AZM within a distance of 4 Å with respect of any atom of the PAMAM for the MMP/AZM-PAMAM systems, the number of hydrogen bond interactions between the PAMAM and the entrapped MMP and AZM molecules.
G4-Asn10 models on a box containing explicit water molecules and Cl- ions. The dendrimer structures are shown by space-filling representation with carbon, nitrogen and oxygen atoms shown in green, blue and red, respectively. The Asn end groups are shown in purple. The Cl- is shown in brown. The chemical structures of MMP and AZM are shown in detail in the center. The distribution of 50 of these molecules in the water box are shown as spacefilling representation with carbon, nitrogen, oxygen, phosphorous and sulfur atoms shown in gray, blue, red, orange and yellow, respectively.
Building molecular structures: The molecular structures of MMP and the representative units of six functionalized PAMAM (monomer-FA, monomer-Cou, monomer-Arg, monomer-Lysine, monomer-Asparagine and monomer- Amine) were built using GaussView program.3838 Dennington, I. I. R.; Keith, T.; Milliam, J.; Eppinnett, K.; Hovell, W. L.; Gilliland, R.; GaussView, Version 3.09, Semichem Inc.: Shawnee Mission, 2003. The geometry of these molecules was optimized at density functional theory (DFT) level using B3LYP method with 6-31G* as the basis set, which has been implemented in Gaussian 03 package program.3939 Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 03, Revision C. 02, Gaussian, Inc.: Wallingford, CT, 2004.
Calculation of interaction energies by nanoinformatic methodologies: Metropolis Monte Carlo simulations2727 Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.; Teller, E.; J. Chem. Phys. 1953, 21, 1087. were used to calculate the interaction energy between conformational pairs [molecule(1)-molecule(2)], in which the molecule(1) represents the monomeric unit of a specific PAMAM attached to a functional group and the molecule(2) represents the OP compound MMP.
The algorithm that generates the conformational sampling includes excluded-volume constraint method.2525 Fan, C. F.; Olafson, B. D.; Blanco, M.; Hsu, S. L.; Macromol. 1992, 25, 3667. Procedures used to calculate the interaction energy were as follows: (i) the mass centers of molecule(1) and molecule(2) are placed near the origin of the Cartesian coordinates frame, (ii) then, the new orientation of molecule(2) [in relation to molecule(1)] is chosen according to a set of three Euler angles (α, β, γ) obtained randomly, (iii) molecule(2) is then translated along the random vector “n” until the vdW surfaces of each molecule touch each other, (iv) after translation, single-point energy (1SCF) for this specific molecular conformation [molecule(1)-molecule(2)] is calculated using a semi-empirical quantum mechanical method (PM6‑DH+)1919 Stewart, J. J.; J. Mol. Model. 2007, 13, 1173. implemented in MOPAC2009TM packaged program, version 11.038L (LINUX),2929 Stewart, J. J. P.; MOPAC2009, Version 10.040L; Stewart Computational Chemistry: Colorado Springs, USA, 2009. (v) the heat of formation (ΔHf) is extracted of the previous result and also from its isolated parts. Then, a supermolecular approach was used to obtain directly the interaction energy (ΔEint) for molecule-molecule pairs, as the difference between the energy of the complex “molecule(1)-molecule(2)” and the sum of the energies of its isolated parts. According to the approach, the Gibbs energy molecule-molecule, molecule-polymer or polymer-polymer is defined by equation 2.

The entropy in the polymer systems2525 Fan, C. F.; Olafson, B. D.; Blanco, M.; Hsu, S. L.; Macromol. 1992, 25, 3667. has been shown to have a small contribution in the ΔG, thus the term TΔS is negligible and equation 3 can be used.

The above equation is conveniently used to estimate the interaction energy with a standard quantum chemical procedure. In order to solve the superposition error caused by this approach in equation 3, a highly efficient conformational sampling, according to what is described by MD calculations, the ΔG of mixing polymer-molecule depends on the specific theory or model used. Thus, it was employed PM6-DH+ theory that is based on the Flory-Huggins lattice approximation, which is the simplest and most widely used to calculate the free energy of polymermolecule interactions, ΔG. A general expression through Flory-Huggins theory for the ΔG of a binary system (molecule-molecule, molecule-polymer, or polymerpolymer) can be described as:

where ΔG is the free energy of polymer-molecule per mole of lattice site, ϕ is the volume fraction, and x is the chain length with each repeating unit defined as occupying a lattice site. The Flory-Huggins χ parameter is defined as:

where z is the coordination number of the model lattice, and ΔEint is the energy of formation of an unlike pair (ΔE1,2). According to equation 5, the energy of a particular [1,2] pair is defined as:

Finally, assuming that ΔG = ΔEint (equation 3), the ΔEint described in this work was defined as:
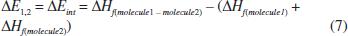
Thus, steps (i) through (v) are repeated until the obtention of 100,000 different molecular configurations and their corresponding interaction energies. Then, from this interaction energies distribution, the average “<ΔE(molecule1 – molecule2)>” was calculated.
The molecule(1) is replaced for each one of the six representative units of the functionalized PAMAM (monomer-FA, monomer-Cou, monomer-Arg, monomer- Lysine, monomer-Asparagine and monomer-Amine) and the above calculations are performed again to finally obtain six-interaction energy averages.
In order to compare and demonstrate the effectiveness of the above methodology, conformations associated with the best interaction energies for each of the 6 analyzed complexes were selected, and energies calculated at DFT level using B3LYP method with 6-31G (d,p) as basis set, which has been implemented in Gaussian 03 package program37. Finally, the interaction energies were calculated based on the supermolecular approach using the equation 7.
AChE inhibition assay
We evaluated in vitro the AChE inhibitory activity of net MMP and encapsulated MMP with G4-Asn and G4 Arg/Lys. The MMP was measured as follows: PAMAM derivatives with MMP were incubated by 45 min on stirring, (molar ratio of MMP:PAMAM = 12:1), after that the AChE inhibitory activity of MMP/PAMAM was measured. Additionally, the AChE inhibitory activity of PAMAM derivatives alone was also evaluated to disregard any AChE inhibitory activity of PAMAM. The assay for measuring AChE inhibitory activity was carried out according to Ellman et al.4040 Ellman, G. L.; Courtney, K. D.; Andres Jr., V.; Feather-Stone, R. M.; Biochem. Pharmacol. 1961, 7, 88. and adapted to 96-well microtiter plates by Lopez et al.4141 Lopez, S.; Bastida, J.; Viladomat, F.; Codina, C.; Life Sci. 2002, 71, 2521. and described by Gutierrez et al.4242 Gutierrez, M.; Theoduloz, C.; Rodriguez, J.; Lolas, J. M.; Schmeda-Hirschmann, G.; J. Agric. Food Chem. 2005, 53, 7701. The enzyme was obtained from electric eel (C-3389, type VI‑S, Sigma Chemical Co., St. Louis, MO). In brief, 50 μL of the sample dissolved in phosphate buffer (8 mmol L-1 K2HPO4, 2.3 mmol L-1 NaH2PO4, 150 mmol L-1 NaCl, and 0.05% Tween 20 at pH 7.6) and 50 μL of the AChE solution (0.25 unit mL-1) in the same phosphate buffer were added to the wells. Then, the plates were incubated for 30 min at room temperature. After the incubation time, 100 μL of the substrate solution [40 mmol L-1 Na2HPO4, 0.2 mmol L-1 5,5'-dithio-bis (2-nitrobenzoic acid) (DTNB), and 0.24 mmol L-1 acetylthiocholine iodide (ACTI) in distilled water at pH 7.5] was added. The absorbance was read in a Bio-Tek Instrument microplate reader at 405 nm after 3 min. The enzyme activity was calculated as a percentage compared to a control using only the buffer and enzyme solution. The compounds were assayed at 100 μg mL-1, and the alkaloid galanthamine was used as the reference compound. When the enzyme inhibition was > 50% at 100 μg mL-1, dilutions were performed to determine the corresponding IC50 values. The IC50 values were calculated by means of regression analysis from three individual determinations.
Results and Discussion
Synthesis of PAMAM derivatives
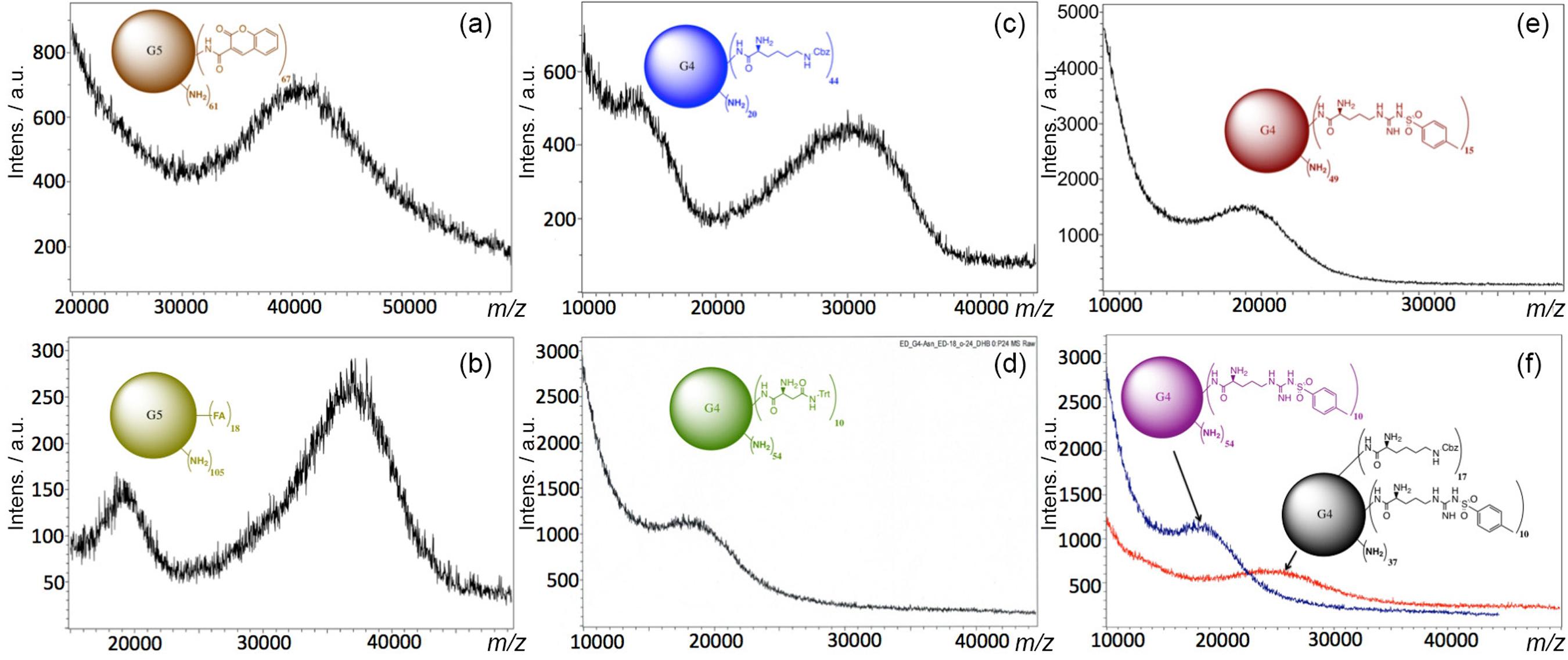
FA and Cou were attached to the amine terminal groups of PAMAM-G5 by EDC·HCl and HOBt coupling reaction.1212 Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem. 2011, 400, 483.,3939 Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 03, Revision C. 02, Gaussian, Inc.: Wallingford, CT, 2004. An excess amount of reagents (1:140) was used to allow functionalization of the 128-terminal amine groups of the PAMAM-G5 (MW ca. 28826) in order to ensure the maximum functionalization. However, only 18 FA (MW of PAMAM G5 + 18 FA ca. 36800) and 67 Cou molecules (MW of PAMAM G5 + 67 Cou ca. 41600) were found attached to the G5, respectively. It is proposed that the difference in the number of molecules coupled to G5 was due to steric hindrance caused by the size of FA and Cou, respectively (Figures 2a and 2b).
MALDI-TOF analysis of (a) G5-Cou67; (b) G5-FA18; (c) G4-Lys44; (d) G4-Asn10 (e) G4-Arg15; and (f) G4-Lys25-Arg14.
Lys, Arg and Asn were coupled to the terminal amines of the G4 by EDC·HCl and HOBt coupling reaction.1212 Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem. 2011, 400, 483.,3939 Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 03, Revision C. 02, Gaussian, Inc.: Wallingford, CT, 2004. An excess amount of reagent (1:70) was used in all three cases to allow maximum possible functionalization. Nevertheless, only 44 molecules of Lys were conjugated to PAMAM (MW G4 + 44 Lys ca. 30500), 15 molecules of arginine were conjugated to G4 (MW G4 + 15 Arg ca. 19800), and 10 molecules of Asn were found attached to the G4 (MW G4 + 10 Asn ca. 18500). Again, it is proposed that the specific number of molecules attached to G4 is determined by the steric hindrance and physicochemical behavior of Arg, Lys and Asn, respectively (Figures 2c-e).
The surface functionalization of PAMAM-G4 with two different amino acids (Arg and Lys) was also achieved. In this case, a small amount of arginine (1:5) was used to accommodate second amino acid lysine by subsequent reaction. With this simple methodology, coupling of two different amino acids on the surfaces of PAMAM-G4 was obtained. The goal of this approach was to increase the capacity of trapping to the OP (synergy) by PAMAM derivatives through more hydrogen bonds, weak interactions such as vdW, hydrophobic bonds, and electrostatic interactions. Thus, it was possible to conjugate 14 molecules of Arg (MW ca. 18600) to G4, and additionally introduce 25 molecules of Lys (MW ca. 25200) (Figure 2f).
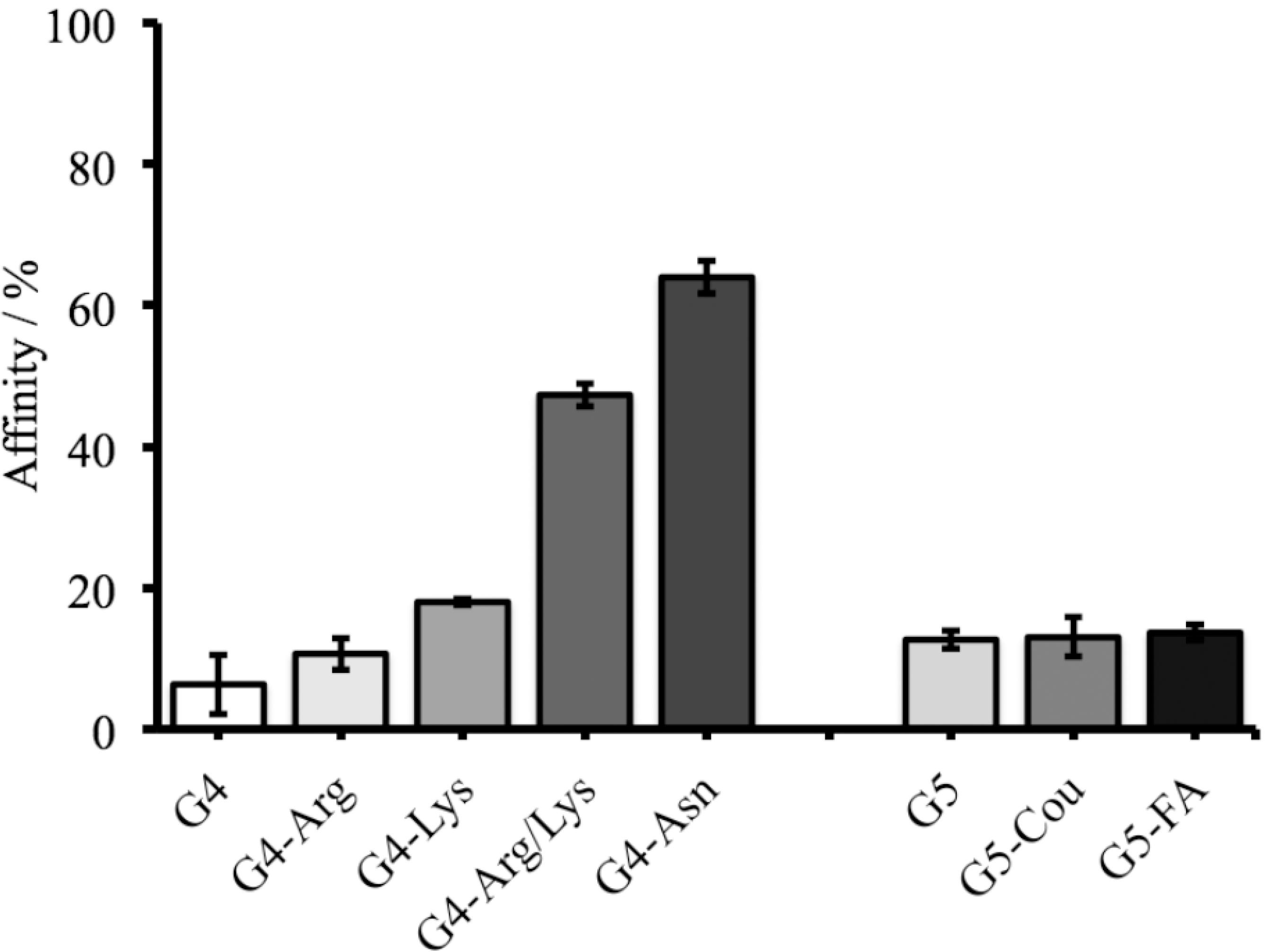
The adsorption behavior of OPs compounds (specifically MMP by PAMAM derivatives) in model solutions was studied through HPLC analysis, but the desorption behavior was not performed. (Figure S1, SI section). It was observed that the adsorption of MMP was higher when it was exposed to G4-Arg/Lys and G4-Asn achieving 45% and 64% of affinity, respectively (Figure 3). This high affinity may be due to the small size of MMP and multiple hydrogen bonds that occur between the tertiary amine and phosphoryl group from MMP and secondary, tertiary amines (interdendritic) and carbonyl groups of PAMAM derivatives (Figure 4). Molecular dynamics calculations can predict and evaluate the contribution of other energies involved in the dendrimer-OP system. In this system, the hydrophobic interaction strength is obtained through the vdW energy. The variation of the vdW energy for the MMP-PAMAM and AZM‑PAMAM systems, are shown in Figure S2 (SI section). The vdW energy pattern do not change significantly with time, it confirms that vdW interaction is less significant than the hydrogen bonding interaction. Another point to consider is that G4-Asn provides greater amount of carbonyl groups to the system than other polymers studied here, allowing the formation of more hydrogen bonds between the PAMAM and the OP.
Experimental assays indicating the percentage of PAMAM derivatives affinity to MMP. As control, G4-NH2 and G5-NH2 were used.
Representation view of the interaction between MMP and G4-Asn indicating the role of hydrogen bond in the capture.
Bioinformatic studies
Six, three-dimensional atomic models for monomer-end-group complexes were generated using the in silico design. These six functionalized PAMAM representative structures were used to estimate the PAMAM/OPs interactions.
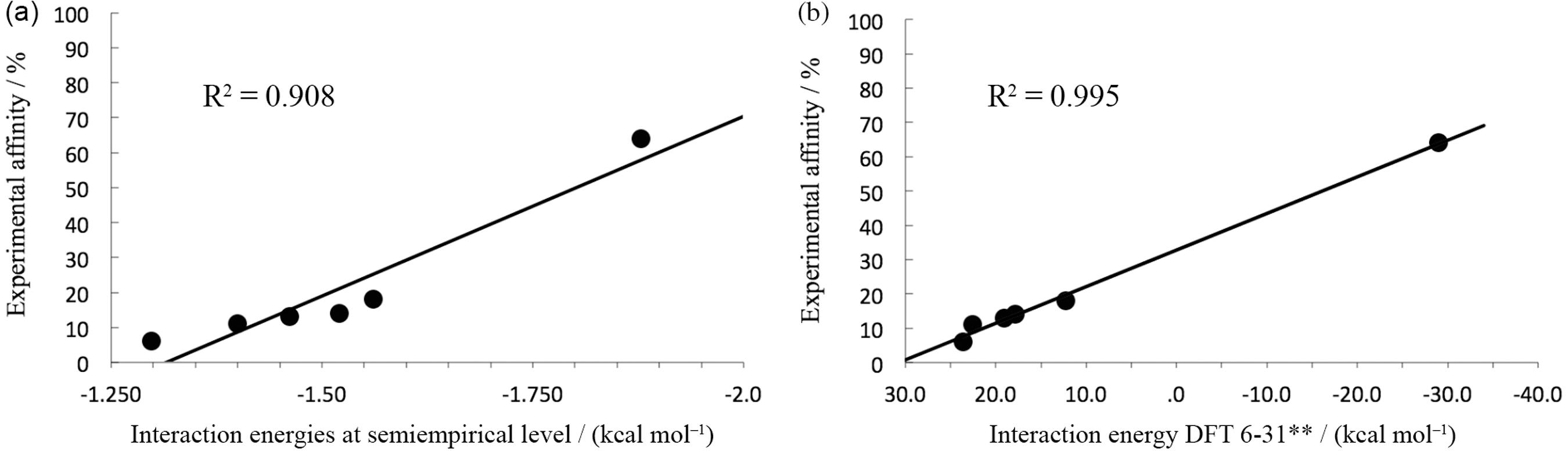
The average interaction energy calculated by the nanoinformatic methodologies is depicted in Table 2. The theoretical data was correlated with the experimental percentage of affinity calculated by HPLC (Figure 5). For the six representative units of the studied functionalized PAMAM, the relative order of affinity by MMP was predicted as: monomer-Asparagine > monomer-Lysine > monomer-Folate > monomer-Coumarin > monomer-Arginine > monomer-Amine. As shown in Table 2 and Figure 5b, interaction energies calculated through DFT‑6‑31G method showed a better correlation (r2 = 0.995) as expected, corroborating the data obtained at semiempirical level. Furthermore, comparing DFT and PM6-FH+ correlations (r2), it can be noted that semi-empirical method gave an acceptable and highly predictive correlation. Moreover, the data obtained by DFT showed to be much more accurate than semi-empirical methods, however, considering the efficiency and speed required for filtering several of thousand conformations, the semi-empirical method probed to be commendable.
Values of the theoretical interaction energies (at semi empirical and DFT level) and experimental (%) affinities between MMP and the monomeric unit of PAMAM attached to six different functional groups
According to the obtained results, the functionalized PAMAM improved its ability to trap OP molecules; thus, strategic changes of the functional groups on the surface of PAMAM have proven to be relevant when defining their interaction with molecules of interest.
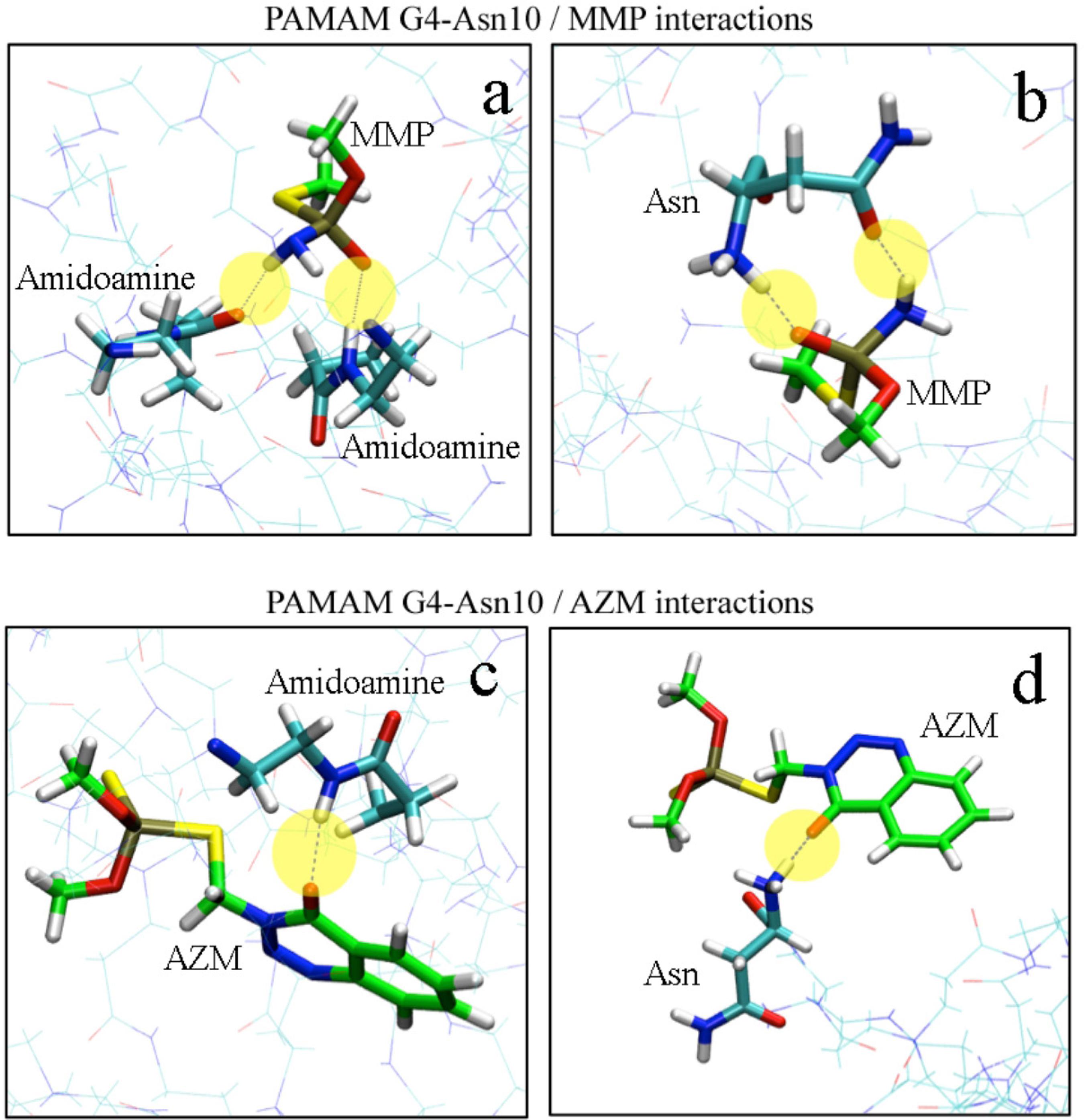
Computational chemistry approaches through nanoinformatics platforms have proved to be essential in the estimation of the PAMAM-molecule interactions, due to the speedy results, the low cost of in silico experiments (this is particularly true for those scenarios where the handling of toxic or otherwise hazardous materials is necessary) and the ability to relate the theoretical values with experimental data with high accuracy (Figure 5). To characterize the G4-Asn10 models and their interaction with MMP and AZM molecules at molecular level, we performed MD simulations to subsequently make a structural analysis of the interactions present in the system. The first analysis performed was the radius of gyration (Rg) calculated as a function of simulated time, as shown in Figure S3 (see SI section). Furthermore, the mean radius of gyration (Rg) was calculated including only the last 8 ns of the 20 ns equilibration simulations, where appeared to take on a steady value. For G4-Asn10 with MMP molecules, values of 21.0 ± 0.2 Å were observed, while for G4-Asn10 with AZM molecules, 21.6 ± 0.3 Å. The second analysis was RDF, which represents the probability to find an atom in a shell (dr) at the distance r, of another atom or molecule. Figure S4 (SI section) shows RDF of G4-Asn10/MMP (1:50) and G4-Asn10/AZM (1:50) systems. The RDF shows a radius r = 4 Å as the distance from which the highest number of MMP molecules are found, whereas for AZM molecules, the radius was higher and lower its probability, radius r = 6 Å and r =10 Å for AZM molecules. Therefore, the MMP molecules are positioned at locations closer to PAMAM than AZM molecules, allowing greater interaction with the terminal groups of G4-Asn10. Distances were calculated between phosphorus atoms (P) of MMP/ AZM molecules and nitrogen atoms (N) of terminal amines and Asn functional groups. The representation of hydrogen bonds between MMP/AZM and G4-Asn10 are shown in Figure 6. The AZM molecule can only form one hydrogen bond with the G4-Asn10 monomers, and the majority of the contributions come from the hydroxyl oxygen atom not associated with the phosphate group. On the other hand, a MMP molecule can achieve two hydrogen bonds with G4-Asn10, the hydroxyl oxygen atom from phosphate group and hydrogen atoms from amine group. Thus G4‑Asn10 can produce more hydrogen bonds in the presence of MMP molecules than AZM, for instance, in the Figure S5 (SI section) is depicted the number of hydrogen bonds as functions of time for MMP and AZM molecules interacting with G4-Asn10. The higher number of hydrogen bonds is generated in the interaction between G4-Asn10 with MMP molecules. The capability of entrapment of G4-Asn10 was calculated through the number of molecules near the PAMAM at a distance of 4 Å or less (value obtained from the RDF analysis). The number of MMP trapped by G4‑Asn10 was higher than AZM: approximately 18‑20 MMP versus 10‑12 AZM molecules in the last 4 ns of the 20 ns equilibration simulation, in the Figure S6 (available as SI) is depicted the number of MMP and AZM trapped by functionalized PAMAM. Thus, it was rationalized that the entrapment of these molecules was promoted primarily by hydrogen bonds formation, especially by of the MMP molecules ability to form more hydrogen bonds with G4‑Asn10.
Correlation between the experimental affinity values obtained by HPLC studies: (a) the interaction energy averages calculated by using semiempirical method (MOPAC); and (b) the interaction energies calculated by DFT (B3LYP/6-31G).
Intermolecular hydrogen bonds (yellow circles) for MMP with G4 (a) and G4-Asn10 (b), intermolecular hydrogen bonds (yellow circles) for AZM with G4 (c) and G4-Asn10 (d).
AChE inhibition assay of PAMAM derivatives
To give further support to our model studies for capturing affinity and molecular simulations data, the best PAMAM derivatives (G4-Asn and G4-Arg/Lys, according to percentage of affinity) were employed to evaluate the inhibition of the AChE activity in vitro. Initially, the inhibition activity of MMP to AChE was measured, obtaining an IC50 of 0.85 μg mL-1, confirming the higher toxicity of MMP. Next, MMP was exposed (incubated) to PAMAM derivatives and G4-NH2 (commercial) for a period of 45 min with constant stirring (molar ratio MMP:PAMAM = 12:1). IC50 values for commercial PAMAM, MMP and derivatized PAMAM are given in Table 3. In the case of MMP trapped by G4-NH2, the data obtained indicated that the IC50 increased slightly, showing IC50 values of 4.23 μg mL-1. Whereas the MMP trapped by PAMAM derivatives reached IC50 values of 65 μg mL-1 and 132 μg mL-1 for G4-Asn and G4-Arg/Lys, respectively. Therefore, the IC50 of MMP encapsulated PAMAM derivatives was significantly higher than MMP encapsulated PAMAM-NH2 and free MMP (see Table 3). Finally, the toxicity of PAMAM derivatives was evaluated (G4-Asn and G4-Arg-Lys). In both cases, the values were higher than 300 μmol L-1 (see Table 3), demonstrating that the toxicity of PAMAM is not significant. These data indicate that PAMAM derivatives have the capacity to capture MMP efficiently, and perhaps, could be used in a near future in preventing the poisoning events. Molecular simulation calculations performed in this study predict that there is a good “hydrogen bond” formation and “radius of gyration” between G4-Asn with MMP. This is consistent with the AChE activity assay, where the IC50 of MMP increased significantly when coupled with PAMAM derivatives. We hypothesize it is due to a higher MMPPAMAM union, due to the formed hydrogen bonds and the closeness of the atoms in each of the molecules involved in the interaction system. Furthermore, if we analyze the in vitro experimental data obtained in the AchE activity assay, we could conclude that the interaction MMP-PAMAM is strong, limiting the contact between MMP and AchE enzyme, presumably by the encapsulation of MMP inside of PAMAM.
Conclusion
Functionalization of PAMAM derivatives with different amino acids such as lysine, arginine, asparagine and others biomolecules (coumarin and folic acid) was demonstrated. These PAMAM derivatives were employed to trap AZM and MMP, OP compounds broadly used as pesticides in the agricultural sector. The molecules were designed and modeled using novel bioinformatic tools, and chemically synthesized, (specifically G4-Asn and G4-Arg/Lys), to demonstrate significant affinity for MMP. The results obtained by HPLC in model solutions, support theoretical chemistry through nanoinformatic methodologies to estimate the interaction energies between MMP and six functionalized PAMAM. In silico results have proven to be highly predictive to estimate a priori, a tendency of affinity from a group of MPP by specific PAMAM, and allowed observing that the PAMAM-molecule interactions play an important role for the development of chemical encapsulation and attachment systems. Moreover, by comparing theoretical affinity of PAMAM derivatives with AZM and MMP, it was demonstrated that the better trapping of MMP was produced by PAMAM-Asn primarily due to hydrogen bonds formation, especially by the MMP ability to form more hydrogen bonds with G4-Asn10. Furthermore, our results of affinity by HPLC and molecular dynamics correlated highly with the in vitro enzyme AChE activity assays. The inhibition of AChE by MMP in solution, highly decreased when G4-Arg/Lys and G4-Asn were used compared with commercial PAMAM G4. This information allows us to conclude that PAMAM derivatives G4-Arg/ Lys and G4-Asn are capable of capturing MMP in solution efficiently, and greatly reduce the MMP action in the enzyme and could be used as possible nano-detoxification agents of OPs in the future. In vivo studies for nanodetoxication of OPs are under current investigation.
Acknowledgments
This work was supported by a grant from E. Durán and L. S. Santos FONDECYT (1140642 and Postdoctoral Grant No. 3120178) and Innova Chile CORFO Code FCRCSB 09CEII-6991. The authors acknowledge support from Proyecto Anillo Cientifico ACT1107. Fabian Avila thanks for doctoral scholarship to CONICYT-PCHA/Doctorado Nacional 2013-21130308. Fondecyt 11130087 (A.J.), 11130086 (F.M.N.) and PIEI (Química y Bio-orgánica en Productos Naturales) from UTalca are also acknowledged.
References
-
1Sogorb, M. A.; Vilanova, E.; Toxicol. Lett. 2002, 128, 215.
-
2Obare, S. O.; De, C.; Guo, W.; Haywood, T. L.; Samuels, T. A.; Adams, C. P.; Masika, N. O.; Murray, N. O. D. H.; Anderson, G. A.; Campbell, K.; Fletcher, K.; Sensors (Basel) 2010, 10, 7018.
-
3Singh, B. K.; Nat. Rev. Microbiol 2009, 7, 156.
-
4Emerick, G. L.; DeOliveira, G. H.; Oliveira, R. V.; Ehrich, M.; Toxicology 2012, 292, 145.
-
5Saheli, P.; Indrani, R.; Gopal, L.; Prasun, P.; Samrat, R.; Arunava, S.; Arunava, G.; J. Environ. Sci. Health, Part B 2013, 48, 559.
-
6Montesano, M. A.; Olsson, A. O.; Kuklenyik, P.; Needham, L. L.; Bradman, A.; Barr, D. B.; J. Exposure Sci. Environ. Epidemiol. 2007, 17, 321.
-
7Sogorb, M. A.; Vilanova, E.; Carrera, V.; Toxicol. Lett. 2004, 151, 219.
-
8Delfino, R. T.; Ribeiro, T. S.; Figueroa-Villar, J. D.; J. Braz. Chem. Soc. 2009, 20, 407.
-
9Bird, S. B.; Sutherland, T. D.; Gresham, C.; Oakeshott, J.; Scott, C.; Eddleston, M.; Toxicology 2008, 247, 88.
-
10Esfand, R.; Tomalia, D. A.; Drug Discovery Today 2001, 6, 427.
-
11Navarro, G.; de ILarduya, C. T.; Nanomed. Nanotechnol. Biol. Med 2009, 5, 287.
-
12Geraldo, D. A.; Durán-Lara, E. F.; Aguayo, D.; Cachau, R. E.; Tapia, J.; Esparza, R.; Yacaman, M. J.; Gonzalez-Nilo, F. D.; Santos, L. S.; Anal. Bioanal. Chem. 2011, 400, 483.
-
13Dong, Z.; Katsumi, H.; Sakane, T.; Yamamoto, A.; Int. J. Pharm 2010, 293, 244.
-
14Devarakonda, B.; Hill, B. R. A.; de Villiers, M. M.; Int. J. Pharm 2004, 284, 133.
-
15Domanski, D. M.; Klajnert, B.; Bryszewska, M.; Bioelectrochem 2004, 63, 189.
-
16Jaga, K.; Dharmani, C.; Rev. Panam. Salud Publica 2003, 14, 171.
-
17Kamanyire, R.; Karalliedde, L.; Occup. Med-Oxford 2004, 54, 68.
-
18Lima, C. S.; Dutra-Tavares, A. C.; Nunes, F.; Nunes-Freitas, A. L.; Ribeiro-Carvalho, A.; Filgueiras, C. C.; Manhaes, A. C.; Meyer, A.; Abreu-Villaca, Y.; Toxicol. Sci 2013, 134, 125.
-
19Stewart, J. J.; J. Mol. Model 2007, 13, 1173.
-
20Korth, M.;J. Chem. Theory Comput 2010, 6, 3808.
-
21Gonzalez-Nilo, F.; Perez-Acle, T.; Guinez-Molinos, S.; Geraldo, D. A.; Sandoval, C.; Yevenes, A.; Santos, L. S.; Laurie, V. F.; Mendoza, H.; Cachau, R. E.; Biol. Res 2011, 44, 43.
-
22Maojo, V.; Fritts, M.; de la Iglesia, D.; Cachau, R. E.; Garcia-Remesal, M.; Mitchell, J. A.; Kulikowski, C.; Int. J. Nanomed. 2012, 7, 3867.
-
23Maojo, V.; Fritts, M.; Martin-Sanchez, F.; de la Iglesia, D.; Cachau, R. E.; Garcia-Remesal, M.; Crespo, J.; Mitchell, J. A.; Anguita, A.; Baker, N.; Barreiro, J. M.; Benitez, S. E.; de la Calle, G.; Facelli, J. C.; Ghazal, P.; Geissbuhler, A.; Gonzalez‑Nilo, F.; Graf, N.; Grangeat, P.; Hermosilla, I.; Hussein, R.; Kern, J.; Koch, S.; Legre, Y.; Lopez-Alonso, V.; Lopez-Campos, G.; Milanesi, L.; Moustakis, L.; Munteanu, C.; Otero, P.; Pazos, A.; Perez-Rey, D.; Potamias, G.; Sanz, F.; Kulikowski, C.; Comput. Sci. Eng. 2012, 94, 521.
-
24de la Iglesia, D.; Maojo, V.; Chiesa, S.; Martin‑Sanchez, F.; Kern, J.; Potamias, G.; Crespo, J.; Garcia-Remesal, M.; Keuchkerian, S.; Kulikowski, C.; Mitchell, J. A.; Methods Inf. Med 2011, 50, 84.
-
25Fan, C. F.; Olafson, B. D.; Blanco, M.; Hsu, S. L.; Macromol 1992, 25, 3667.
-
26Avila-Salas, F.; Sandoval, C.; Caballero, J.; Guinez‑Molinos, S.; Santos, L. S.; Cachau, R. E.; Gonzalez-Nilo, F. D.; J. Phys. Chem. B 2012, 116, 2031.
-
27Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.; Teller, E.; J. Chem. Phys 1953, 21, 1087.
-
28França, T. C.; Guimarães, A. P.; Cortopassi, W. A.; Oliveira, A. A.; Ramalho, T. C.; Curr. Comput.-Aided Drug Des. 2013, 9, 507.
-
29Stewart, J. J. P.; MOPAC2009, Version 10.040L; Stewart Computational Chemistry: Colorado Springs, USA, 2009.
-
30Pisal, D. S.; Yellepeddi, V. K.; Kumar, A.; Kaushik, R. S.; Hildreth, M. B.; Guan, X.; Palakurthi, S.; Int. J. Pharm 2008, 350, 113.
-
31Durán-Lara, E. F.; López-Cortés, X. A.; Castro, R. I.; Ávila-Salas, F.; González-Nilo, F. D.; Laurie, F. V.; Santos, L. S.; Food Chem. 2015, 168, 464.
-
32Phillips, J. C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R. D.; Kale, L.; Schulten, K.; J. Comput. Chem 2005, 26, 1781.
-
33Zoete, V.; Cuendet, M. A.; Grosdidier, A.; Michielin, O.; J. Comput. Chem. 2001, 32, 2359.
-
34Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L.; J. Chem. Phys 1983, 79, 926.
-
35Feller, S.; Zhang, Y.; Pastor, R.; Brooks, B.; J. Chem. Phys 1995, 103, 4613.
-
36MacKerell Jr., A. D.; Bashford, D.; Bellott, M.; Dunbrack, R. L.; Evanseck, J. D.; Field, M. J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; Joseph-McCarthy, D.; Kuchnir, L.; Kuczera, K.; Lau, F. T. K.; Mattos, C.; Michnick, S.; Ngo, T.; Nguyen, D. T.; Prodhom, B.; Reiher, W. E.; Roux, B.; Schlenkrich, M.; Smith, J. C.; Stote, R.; Straub, J.; Watanabe, M.; Wiórkiewicz-Kuczera, J.; Yin, D.; Karplus, M.; J. Phys. Chem. B 1998, 102, 3586.
-
37Humphrey, W.; Dalke, A.; Schulten, K.; J. Mol. Graphics 1996, 14, 33.
-
38Dennington, I. I. R.; Keith, T.; Milliam, J.; Eppinnett, K.; Hovell, W. L.; Gilliland, R.; GaussView, Version 3.09, Semichem Inc.: Shawnee Mission, 2003.
-
39Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 03, Revision C. 02, Gaussian, Inc.: Wallingford, CT, 2004.
-
40Ellman, G. L.; Courtney, K. D.; Andres Jr., V.; Feather-Stone, R. M.; Biochem. Pharmacol 1961, 7, 88.
-
41Lopez, S.; Bastida, J.; Viladomat, F.; Codina, C.; Life Sci. 2002, 71, 2521.
-
42Gutierrez, M.; Theoduloz, C.; Rodriguez, J.; Lolas, J. M.; Schmeda-Hirschmann, G.; J. Agric. Food Chem 2005, 53, 7701.
-
Supplementary InformationSupplementary data are available free of charge at http://jbcs.sbq.org.br as PDF file.
-
Additions and Corrections
On page 590, where it was read in Acknowledgements section:“This work was supported by a grant from E. Durán and L. S. Santos FONDECYT (Postdoctoral Grant No. 3120178) and Innova Chile CORFO Code FCR-CSB 09CEII-6991. The authors acknowledge support from Proyecto Anillo Científico ACT1107. Fabian Avila thanks for doctoral scholarship to CONICYT-PCHA/Doctorado Nacional 2013-21130308. Fondecyt 11130086 (F.M.N.) and PIEI (Química y Bio-orgánica en Productos Naturales) from UTalca is also acknowledged.”Now read:“This work was supported by a grant from E. Durán and L. S. Santos FONDECYT (1140642 and Postdoctoral Grant No. 3120178) and Innova Chile CORFO Code FCR-CSB 09CEII-6991. The authors acknowledge support from Proyecto Anillo Cientifico ACT1107. Fabian Avila thanks for doctoral scholarship to CONICYT-PCHA/Doctorado Nacional 2013- 21130308. Fondecyt 11130087 (A.J.), 11130086 (F.M.N.) and PIEI (Química y Bio-orgánica en Productos Naturales) from UTalca are also acknowledged.
Data availability
Publication Dates
-
Publication in this collection
Mar 2015
History
-
Received
09 Sept 2014 -
Accepted
03 Feb 2015