
Abstracts
Electrochemical properties of soils are very important for the understanding of the physico-chemical phenomena which affect soil fertility and the availability of nutrients for plants. This review highlights the electrochemical properties of tropical soils, the behavior and the availability of nutrients, toxic elements and heavy metals in the soil, especially for soils with predominant variable charge minerals. Availability of the elements is related to ionic exchange, solution speciation, and electrostatic and specific adsorptive soil properties. Empirical and surface complexation models are briefly described, and some results of their application in tropical soils are presented. A better understanding of the role of the double diffuse layer of charges and CEC on nutrient cation availability for highly weathered soils is required, as well as a solid comprehension of surface complexation models, in order to improve the knowledge regarding the behavior of anions in soils. More studies have to be conducted to generate results that enable the use of chemical speciation concepts and calculation of several constants used in surface complexation models, especially for highly weathered soils from the humid tropics. There has to be a continuing development and use of computer softwares that have already incorporated the concepts of chemical speciation and adsorption models in the study of nutrients, toxic elements and heavy metal availability in the soil-plant system.
electric charge; adsorption; model; ionic exchange; speciation
As propriedades eletroquímicas dos solos tropicais são muito importantes para entendimento dos fenômenos físico-químicos que afetam a fertilidade do solo e a disponibilidade dos nutrientes das plantas. Essa revisão destaca os atributos eletroquímicos de solos e o comportamento e a disponibilidade de nutrientes, elementos tóxicos e metais pesados no solo, especialmente aqueles com predominância de minerais com cargas variáveis. A disponibilidade dos elementos é relacionada com a troca iônica, especiação da solução e propriedades adsortivas eletrostáticas e específicas do solo. Modelos empíricos e de complexação de superfície são brevemente descritos, e são apresentados resultados de sua aplicação em solos tropicais. É necessário um melhor entendimento do papel da dupla camada difusa de cargas e da CTC na disponibildade de nutrientes em solos altamente intemperizados, assim como uma melhor compreensão dos modelos de complexação de superfície, a fim de melhorar o entendimento do comportamento dos ânions no solo. Mais estudos devem ser conduzidos para gerar resultados que tornem possível o uso de conceitos de especiação química e o cálculo de diversas constantes usadas em modelos de complexação, especialmente para solos altamente intemperizados do trópico úmido. Deve haver um contínuo desenvolvimento de programas computacionais que já tenham incorporado os conceitos de especiação e modelos de adsorção no estudo da disponibilidade de nutrientes, elementos tóxicos e metais pesados no sistema solo-planta.
carga elétrica; adsorção; modelo; troca iônica; especiação
REVIEW
Electrochemical attributes and availability of nutrients, toxic elements, and heavy metals in tropical soils
Atributos eletroquímicos e disponibilidade de nutrientes, elementos tóxicos e metais pesados em solos tropicais
Mauricio Paulo Ferreira FontesI,* * Corresponding author < mpfontes@ufv.br> ; Luís Reynaldo Ferracciú AlleoniII
IUFV/Depto. de Solos. Av. P.H. Rolfs, s/n. - 36570-000 - Viçosa, MG - Brasil.
IIUSP/ESALQ - Depto. de Ciência do Solo. CP 09 - 13418-900 - Piracicaba, SP - Brasil.
ABSTRACT
Electrochemical properties of soils are very important for the understanding of the physico-chemical phenomena which affect soil fertility and the availability of nutrients for plants. This review highlights the electrochemical properties of tropical soils, the behavior and the availability of nutrients, toxic elements and heavy metals in the soil, especially for soils with predominant variable charge minerals. Availability of the elements is related to ionic exchange, solution speciation, and electrostatic and specific adsorptive soil properties. Empirical and surface complexation models are briefly described, and some results of their application in tropical soils are presented. A better understanding of the role of the double diffuse layer of charges and CEC on nutrient cation availability for highly weathered soils is required, as well as a solid comprehension of surface complexation models, in order to improve the knowledge regarding the behavior of anions in soils. More studies have to be conducted to generate results that enable the use of chemical speciation concepts and calculation of several constants used in surface complexation models, especially for highly weathered soils from the humid tropics. There has to be a continuing development and use of computer softwares that have already incorporated the concepts of chemical speciation and adsorption models in the study of nutrients, toxic elements and heavy metal availability in the soil-plant system.
Key words: electric charge, adsorption, model, ionic exchange, speciation
RESUMO
As propriedades eletroquímicas dos solos tropicais são muito importantes para entendimento dos fenômenos físico-químicos que afetam a fertilidade do solo e a disponibilidade dos nutrientes das plantas. Essa revisão destaca os atributos eletroquímicos de solos e o comportamento e a disponibilidade de nutrientes, elementos tóxicos e metais pesados no solo, especialmente aqueles com predominância de minerais com cargas variáveis. A disponibilidade dos elementos é relacionada com a troca iônica, especiação da solução e propriedades adsortivas eletrostáticas e específicas do solo. Modelos empíricos e de complexação de superfície são brevemente descritos, e são apresentados resultados de sua aplicação em solos tropicais. É necessário um melhor entendimento do papel da dupla camada difusa de cargas e da CTC na disponibildade de nutrientes em solos altamente intemperizados, assim como uma melhor compreensão dos modelos de complexação de superfície, a fim de melhorar o entendimento do comportamento dos ânions no solo. Mais estudos devem ser conduzidos para gerar resultados que tornem possível o uso de conceitos de especiação química e o cálculo de diversas constantes usadas em modelos de complexação, especialmente para solos altamente intemperizados do trópico úmido. Deve haver um contínuo desenvolvimento de programas computacionais que já tenham incorporado os conceitos de especiação e modelos de adsorção no estudo da disponibilidade de nutrientes, elementos tóxicos e metais pesados no sistema solo-planta.
Palavras-chave: carga elétrica, adsorção, modelo, troca iônica, especiação
1. INTRODUCTION
Electrochemical properties of soils are very important for the understanding of the physico-chemical phenomena which affect the availability of nutrients for plants, and, therefore, soil fertility. Defined as the capacity of the soil to supply crops with essential elements, soil fertility is a very complex matter, be cause it is the result of the combination of various processes that occur in the soil and dependent upon several soil attributes such as, texture, structure, content and type of clay minerals, content and type of organic matter (OM), nature and transformations of the parent material, for example.
Nutrient availability or bioavailability is closely connected to soil fertility. Bioavailability is affected by different features and soil characteristics such as dense or compact layers, nutrient diffusion, oxygenated roots and so on. Bioavailability of essential elements can also be defined in mechanistic terms (Sposito, 1989): the element is bioavailable if it is in a chemical form that the plant is able to absorb promptly, and if absorbed, it affects the life cycle of the plant. In the view of Soil Chemistry, bioavailability of an element is determined by competition among plant roots, soil solution and solid phases of soil. The electrochemical properties of the solid phase influences directly the behavior of soil elements, especially in both soil solution composition and in the bioavailability of these elements.
In certain soils, like those with predominant constant charge minerals, the two properties most related to the control of movement and to the immobilization of nutrient elements, are the specific surface area (SSA) and surface charge density. These properties altogether define the cation exchange capacity (CEC). In these soils, which present the majority of their charges as permanent, the CEC and the percentage of base saturation (V%) are the most frequent parameters to evaluate soil fertility. These two parameters vary with other soil properties, such as pH and water holding capacity, and in this way, when used in a combination, they allow good inferences on the soil capacity to supply nutrients to plants.
On the other hand, in soils which present predominant variable charge minerals, the properties most directly related to nutrient availability should include the kind of the exposed surface of clay minerals, altogether with the specific surface area and the surface charge density. The value of CEC, V% and the properties that vary with them, are not, in general, good enough to make a sound interpretation of the bioavailability of the nutrients, that is, of soil fertility.
The main reactions which affect the availability of soil nutrients are the losses either by volatilization or leaching, precipitation and adsorption. Few elements are volatile, and the majority does not reach enough concentrations in soil solutions to cause their precipitation. Therefore, reactions related to nutrient availability are the losses by leaching and adsorption, which are directly influenced by electrochemical soil properties.
This review has the objective of relating the electrochemical properties of soils with the availability of nutrients for plants, especially for soils with predominant variable charge minerals, whose importance is emphasized in the humid tropics. The state of art in this field is discussed in a tentative to outline the perspectives for future studies related to soil electrochemical properties and their influence in the availability of nutrients, toxic elements and heavy metals in the soil-plant system.
2. INTERFACE SOLID - SOLUTION
2.1 Charge on soil minerals
The surfaces of soil colloidal materials are electrically changed, in other words, the surface has an excess or a deficit of electrons (Uehara & Gillman, 1981) that compose a double layer (DL) of charges with the ions in soil solution (van Olphen, 1977). If the DL of charges results from internal deflects and structural isomorphic substitutions, the charge density is constant, while the particle surface electric potential varies. If the DL is created by adsorption of potential determining ions (PDI), which are able to modify colloid charges), the density of charge varies, while the surface electric potential is constant and determined solely by the concentration or activities of the ions in solution.
2.1.1 Permanent charges
Permanent charges, also called structural charges, result from isomorphic ionic substitutions in mineral structures and are present at any soil pH. As a consequence, the surface charge of many soil minerals is pH-dependent. Isomorphic substitutions occur in primary minerals during their magmatic formation, while it occurs in secondary minerals either in their soil transformations or by direct inheritance of primary minerals. However, they produce significant charge only in 2:1 clay minerals (smectites and vermiculites) and in a few primary minerals, such as mica, when they have a size small enough to compose the clay fraction of soils.
2.1.2 Variable charges
Variable charges originate from ion adsorption on soil colloid surfaces having the net charge determined by one ion which is highly adsorbed. PDI in soil solution are H+ and OH-, and we can classify the main particles electrically charged in soils as "colloids with pH dependent charges". Sine some other ions can also act as PDIs, such as phosphates, silicates and heavy metals; colloids with variable charge should be the best definition (Uehara & Gillman, 1981). Kaolinite, goethite, hematite and gibbsite, found predominantly in Brazilian soils (Fontes & Weed, 1991), are the main soil minerals with variable charge.
In tropical environments, high temperatures and high rainfall provide appropriate conditions for the rupture of primary and secondary mineral structures and the formation of simpler compounds, such as iron and aluminum oxides/hydroxides. Different from ionic substitution, the charges in these oxides and hydroxides are highly pH-dependent. In these compounds, the more acid the medium, the higher the probability of protonation on the surface (H+ adsorption). In this case, the oxide or the hydroxide will have a positive surface charge and will be able to retain anions such as nitrates, sulphate, chlorides etc. On the other hand, as soon as pH increases, the O-H bond becomes weaker, and H can be released into the soil solution, then generating negative charges. In this specific situation, the surface will retain cations, such as Ca, Mg, and K. This can happen, for example, on Al hydroxide surfaces, that may act either as anion or as cation exchangers. In subsurface horizons of highly weathered soils with low OM contents, Fe and Al oxides may prevail and compose the main surface of ionic exchange. Depending on the pH, the anion exchange capacity (AEC) may surpass the cation excharge capacity (CEC). Large areas in the North of the São Paulo state, South of the Minas Gerais state and Central Brazil present these soils.
On the surface of OM colloids, the variable charge is negative and is originated from the dissociation of hydroxyl on carboxylic, phenolic, enolic groupings etc. Some groupings dissociate at low pH values, whereas others at high pHs. So, negative charges take a place at a great pH range, providing a great buffer capacity in soils with high OM contents. Under pHs commonly found in humid tropical soils, due to certain characteristics, positive charges on OM components are less likely to occur.
2.2 Surface functional groups
An essential characteristic to formation and development of charges in colloids with variable charges is the possibility of surfaces becoming hydrolyzed. In water, the ions Si, Al or Fe, for example, located on certain mineral surfaces, such as kaolinite, hematite, goethite and gibbsite, complete their coordination layers with hydroxyl ions, and their surfaces become hydrolyzed (Stumm, 1992). Positive or negative charges may be created (Parks & de Bruyn 1962; Herbilon, 1988) through hydroxyl protonation or disprotonation (Figure 1), for oxides at an octahedrical configuration and a trivalent metal.
Hydroxyl grouping in this mineral might be connected to one, two, or even three metals, according to Russel et al., (1974) and Parfit et al. (1975). Therefore, they can be named as simple, double and triple hydroxyl coordination. Figure 1 shows extreme protonation cases, with a maximum of positive charges, and disprotonation, with maximum negative charges. Between the extremes, there is a point where the charge is 0 (zero), either due to the absence of charge or to the equal amounts of positive and negative charges. This point is called zero point of charge (ZPC).
3. ADSORPTIVE SOIL PROPERTIES VERSUS NUTRIENTS AVAILABILITY
A previous knowledge of soil properties helps understanding both phytoavailability and metal mobility. When entering the soil, the solute may be either be quickly adsorbed on closer surfaces, or slowly adsorbed by spreading out through the micropores, as soon it reaches distant surfaces or intracrystalline sites (Harter & Naidu, 2001). Several mechanisms are involved in adsorption such as non-specific adsorption, specific adsorption and complexation with soil OM (Camargo et al., 2001). According to Harter & Naidu (2001); interactions between heavy metals and soil particles are non-specific, presenting a small fraction adsorbed by chemisorption.
Apparent irreversibility of adsorbed reactions may be granted to metastable state of surface reactions and allows ionic species elements to be desorbed, causing secondary precipitation reactions (Ford et al., 2001). The precipitation of heavy metals only occurs when they have high concentrations, after the soil solids are dissolved to form soluble constituents.
Electrostatic attraction and covalent bonding between chemical elements and colloid particles are very important for soil fertility studies. Electrostatic attraction is fairly weak and not very stable and, in this reaction, the hydration water remains and outer-sphere complexes are formed. Some examples are the outer-sphere complexes formed with calcium, magnesium, potassium, the exchangeable cations and certain anions, such as carbonate, nitrate, and chloride. It is important to emphasize that nitrate is a highly soluble anion and, therefore, it does not have a strong interaction with soil colloids as it happens with other anions (Havlin et al., 1999). Consequently, it may be leached when its content is high in a wet soil. Losses of NO3- by leaching are the main cause of N losses in well drained soils from the humid tropic in irrigated crop systems. The covalent bonding, in turn, is quite different, since the electrons are shared and the hydration water does not remain. The adsorption is, then, called specific and inner-sphere complexes are formed. Some examples of covalent bonding between chemical elements and soil colloids are the polyprotic anions, such as phosphate and silicate, and some heavy metals, such as Cu, Pb, Cd, Zn, and possibly Ni.
Abreu et al. (2005) analyzed B, Cu, Fe, Mn, Zn, Cd, Cr, Pb, and Ni from 13,416 soil samples (0-0.2 m depth) collected in 21 Brazilian states, 58% of them from the state of São Paulo, from 1993 to 1999. There was no control of possible samples having been taken on the same areas in different years. According to the authors, the number of contaminated samples was small. Even so, the specific cases in which the elements were present in high concentrations, with risk of reaching the food chain, deserve attention. They warn that Cd was of particular concern, because of its possible accumulation to potentially harmful levels for humans in the food chain. This element differs markedly from Pb, since it is mobile in plants, whereas lead is not.
To better correlate soil fertility and electrochemical properties of soil colloids, the two types of bonding should be studied in detail. So, the double layer of charges and CEC (electrostatic bonding) and the adsorption models (specific adsorption) will be described as follows:
3.1 Electrostatic adsorption
3.1.1 Double layer of charges
In a solution, colloidal particles are electrically charged and attract ions of opposite charge. This phenomenon will generate an electric double layer of charges with an electric potential y at the solid solution interface. The number of charges on the particle surface, in measured through the density of charges in the particle per unit area, which is used in many models that describe the double layer of charges in colloidal systems. The main models will be now described.
3.1.1.1 Helmholtz Model
It is a very simple model and it was the first description of the double layer of charges. Helmholtz stated that the double layer would act as a capacitor, where the negative charge is distributed all over the surface, and the charges are lined parallel to the surface at a certain distance (x). In this model, the ions that counterbalance the charges of the particle surface remain in the parallel plane, neither separating from the surface, nor getting closer as well. Cation concentration is determined by s, and the anions concentration in the Helmholtz layer is zero.
3.1.1.2 Gouy-Chapman Model
This model was proposed independently in 1910 and 1913 by Gouy and Chapman, respectively, and it is based on the assumption that ion distribution around a charged particle results from the balance among electrostatic strength, responsible for attracting ions of opposite charge to the surface, and the diffusion strength, responsible for ion dispersion. The final product is a diffuse and reciprocal distribution of anions and cations around the electrically charged particles. The concentration of anions and cations in the external solution is named Co and, by approaching a negatively charged surface, the concentration of cations increases, and that of the anions decreases. The excess charge in solution near the charged particle is called surface charge density (s). The electrical potential (j) decreases from a maximum value at the surface of the charged particle (jo) to zero at infinite, following an exponential distribution. In a mathematical description, it is assumed that the particles are evenly distributed all over the surface; the opposite ions do not present any volume and may approach the surface and ions and surface interact without any ionic specificity.
The space charge density defined as (r) equals the number of charges per unit volume as in

Space charge density, in C cm-3, is defined as:

where Ci is the concentration of ion i per cm3; Zi its valency, and F the Faraday constant.
Poisson's equation relates the divergent of the electric potential (j) in the double layer to the space charge density r (equation 3):

where e is the dielectric constant of the solvent.
The ion distribution in the DL is given by Boltzman's equation:

where Ci is the concentration of ion i at a position where the electric potential is y; Coi the concentration of ion i in the external solution; R the gas universal constant, and T the absolute temperature.
By means of mathematical derivations and transformations, two important formulas are finally obtained. Both equations are very important for a better understanding of the DL of charge. The first is:

where K-1 is the parameter related to the "thickness" of the double layer, also called the "gravity center" of the double layer. According to this theory, the thickness of double layer is directly proportional to the solvent dielectric constant and to the temperature, but inversely proportional to the ion valency and to the ion concentration in solution.
The second formula equates s with the ion concentration and with the electric potential at the particle surface:

The use of Chapman's equation to calculate the surface charge density is very restrict, since the early assumptions are not realistic and, therefore, high cation concentrations may be found on particle surfaces, based on the assumptions that ions act punctually and may reach the surface in an unlimited way.
3.1.1.3 Stern Model
In 1924, Stern combined Helmholtz and Gouy-Chapman theories. He formulated a theory in which he divided the solution near the particle surface in two parts. In the layer closest to the surface, the ions form a compact layer of counter ion charge that is similar to the one described in the Helmholtz model, and the second solution layer is the one already outlined by the diffuse layer model developed in the Gouy-Chapman theory. Stern double layer model assumes that the ions have specific interaction energy and are of a finite size. Thus, the ions will reach the surface in a limited way and will be located at some nanometers from the particle surface. Ions which balance the surface charge are mostly in the Stern layer, and the other in the diffuse Gouy-Chapman layer.
Stern layer has a finite capacity to accommodate ions, and the amount of filled sites depends on various factors, such as ion concentration, ion charge, temperature, dielectric constant of the solvent, and specific properties of the ions. Assuming that the sum of the ions in the Stern layer and in the diffuse layer balance the surface charge density, we have:

in which:

where N1 is the number of adsorption sites per area unit, Na the Avogadro number, w the solvent density; M the solvent molecular weight; yd the electric potential in the Stern layer, and F is the specific adsorption potential.
The charge density s2 in the Gouy-Chapman is given by equation 6.
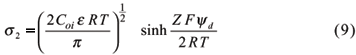
Gouy-Chapman and Stern models were evaluated to describe charge characteristics of an Anionic Acrudox and two Rhodic Acrudoxes of the São Paulo State Brazil (Alleoni & Camargo, 1994a). Samples were collected in regions of large occurrence of these soils, supporting many cash crops. In the highest concentration of the electrolyte (0.1 mol L-1 KCl), the obtained values were better related to the Stern model. On the other hand, under more diluted conditions (KCl 0.01 and 0.001 mol L-1), corresponding to more usual ionic strengths found in highly weathered soils, the variation of surface charge density of the samples with electric potential was in better agreement with the Gouy-Chapman theory.
Wann & Uehara (1978) present an interesting example to illustrate the use of the Stern theory relating fertilization and manipulation of soil electric charges. These authors noticed that in the presence of Ca, the ZPC of an Oxisol decreased from 5.0 to 3.5 after phosphorus application. However, high levels of Ca could revert electric charges when this element was adsorbed on the Stern layer, and this process could cause a reduction for soils retaining in other cations like K, though the electric charge had been increased. Thus, from the soil charge management point of view, the authors recommended triple superphosphate instead of single superphosphate due to its lower Ca/P ratio.
3.1.2 Double layer and Ionic Exchange
Surface charges of soil colloidal particles are balanced by the accumulation of ions of opposite charge and by the reduction of ions of similar charge. Ions which are opposite to soil charged particles are present is excess in the DL and are called exchangeable ions, because any ion can be replaced by other ones, modifying the chemical composition of the electrolyte solution.
In most soils, the predominant charge is negative, and therefore, ions close to particles are the cations, so called exchangeable cations. Maximum number of exchangeable cations in the DL, measured as electric charges per mass unit, under specific solution conditions, corresponds to the CEC. In some cases, either under natural or manipulated conditions, soil colloid surface charge can be predominantly positive. This situation is found in some highly weathered soils, where the variable-charge minerals predominate. Anions electrostatically bound in the DL are called exchangeable anions, and the maximum number of such anions per mass unit is the anion exchange capacity (AEC). However, the most common situation for the majority of soils is the dominance of negative charges over positive charges, and, therefore the most important phenomenon is the CEC.
The deficit of charge density is:

So that the charge density due to the excess or net charge, either in positive or in negative systems, is:
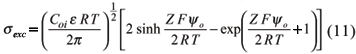
Equation (11) shows that the charge density depends on the electrolyte concentration (Coi) and on the surface electric potential (yo). In a constant charge system, like 2:1 clay minerals, CEC will not change when the electrolyte concentration varies. Nevertheless, at a constant potential system, with predominance of 1:1 clay minerals and Fe and Al oxides, CEC will be strongly affected by the electrolyte concentration. In general terms, on predominant variable charge systems, the charge density due to anion deficit is neglected, and CEC may be taken as the total charge density of DL (equation 6):
For constant potential systems, such as soil minerals, where the potential determining ions are H+ and OH-, the Nernst equation allows the calculation of the potential (van Olphen, 1977; Hunter, 1993)
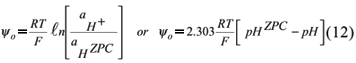
Equation (12) relates the surface potential to the H+ activity, in relation to H+ activity when the surface potential is zero, that is, at the ZPC. Consequently, replacing yo by 2.303 RT/F (pHzpc pH) in equation (6), we have:

In such a system, CEC does not depend only on the electrolyte concentration, but also on the distance between soil pH and the pH at the zero point of charge. Fertilization may affect the charge density of a soil with predominant variable charges. At the same time, electrolyte addition also decreases the surface potential, by changing the solution pH (Uehara & Gillman, 1981).
The more weathered a soil the closer is the tendency of the soil pH to reach its ZPC. The high degree of weathering causes the accumulation of kaolinite, gibbsite, goethite and hematite, besides the amorphous Fe and Al oxides, in the clay fraction of tropical soils (Fontes & Weed, 1991). As a consequence, these soils may have high ZPC in subsurface horizons, the difference between ZPC and soil pH tends to be zero, and the surface charge density tends to be null. Under extreme weathering conditions, soils may have a pH close to 6.0 and very low contents of Ca, Mg, K, and Al.
Alleoni & Camargo (1994b) found values of ZPC varying from 5.6 to 6.0 in Bw horizons of Brazilian Oxisols, showing the large influence of the oxides in the charge balance. Cation retention in this horizon was lower than 3 mmolc kg-1, and the pH measured in 1 mol L-1 KCl was higher than the pH in water, which means, the DpH was positive. The authors also observed that ZPC had positive correlation with the amount of gibbsite and DpH, and negative correlation with the weathering index (Ki). Despite the low fertility, such soils can support a high-level agriculture when correct managed, as it can be noticed in some regions of Northern São Paulo State, Southern Minas Gerais State as well as in Central Brazil (Alleoni & Camargo, 1995). DpH has been greatly used as an indicator of soil net charge (Mekaru & Uehara, 1972). However, some authors point out that this use is only possible for soils with predominant variable charge minerals (Tessens & Shamshuddin, 1982) and DpH ranging from -0.5 and +0.5 (Uehara, 1979). On surface layers of agricultural soils, negative values of DpH are of high occurrence, i.e., pH measured in 1 mol L-1 KCl is usually lower than the pH in H2O.
Managing the fertility of humid tropic soils involves procedures which cause alterations in the soil system and can be discussed based on the equation (13). These situations comprise, for example, increasing electrolyte concentration (Coi) by fertilizer use and also increasing the amount of divalent ions (Ca and Mg) due to liming, which affects the valency (Z) and raises pH. Application of high rates of phosphate fertilizers may cause a decrease in ZPC, due to specific adsorption of phosphate on Fe and Al oxides surfaces (Rajan, 1976). In such cases, the negative net charge rises and may increase Ca, Mg and K retention. In this reasoning, it must be possible to reduce the soil ZPC, particularly in highly weathered soils with low OM contents, by adding specific adsorption anions, such as silicate, that increase the negative surface charge. The use of the silicate has the advantage to increase P bioavailability due to the competition of both anions for the adsorption sites.
In soils with positive net charge, cation and anion movements in the soil profile are affected, and soil fertility is consequently altered. This is remarkable in studies with gypsum, where Ca2+ and SO42- are the predominant ions in soil solution. Camargo & Raij (1989) observed a greater retention of the sulphate ion in an acidulated sample (pH 3.7) of an acric Oxisol with high Fe oxide contents than in the same sample with a greater pH (5.7). These authors pointed out that in the first case the net charge was positive (+27 mmolc kg-1), while it was null at the higher pH.
Uehara & Gillman (1981) cite the example of a high weathered soil in which the soil pH increases even with base losses by leaching, and state that a soil with positive charge balance should have small amounts of exchangeable bases in the subsurface layers, despite the increase of pH with depth. Based on the fact that the nitrate ion (NO3-) can be adsorbed to colloids whose surface is positively charged (Wild, 1972; Wong et al., 1990; Cahn et al., 1992), Hartemink et al. (1996) proved that integration of trees which explore the soil more deeply than annual crops may be more effective in reducing nitrate losses and, therefore, allow its recycling in the soil-plant system.
Generally, CEC is defined in terms of mmol of charge either per unit volume (for soil fertility analysis) or per unit mass (for soil classification). However, CEC can be related to the specific surface area (SSA) and surface charge density (s):

The buffer capacity (BC) of soils with predominant variable charges can be related to the DL theory. Singh & Uehara (1986) presented the BC in these systems as:
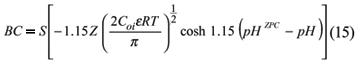
In practice, BC can be expressed as the resistance of soils to pH changes. Soils with high clay and OM levels contain a higher number of adsorption sites when compared to sandy soils with low OM. In these sites, hydrogen is bound in a covalent way, constituting a pool, which is triggered when the H+ level of the solution drops. This is the potential acidity, reported in the results of soil analysis as H+ + Al.
The buffer capacity is related to the doses of lime required for the correction of soil acidity. When lime is added to a high BC soil, there is a natural reposition of H+ by the colloids, to keep the equilibrium constant. Soil potential acidity is effectively neutralized when the pool of covalent H is consumed. In some Brazilian states, like São Paulo, the formula used to calculate rates doses takes into consideration the potential acidity. In this formula, the difference between the desired V% (based on the crop) and the current soil V% is multiplied by the total CEC, which contemplates the potential acidity of the soil.
By the formula of BC, it is seen that the valency of the counterion has a greater influence on the BC than on the CEC. According to Sing & Uehara (1986) this would explain why the lime requirement, determined with NaOH can underestimate the need of CaCO3 to produce a certain variation of pH. These authors also comment that this equation helps to explain a common problem found in tropical soils, that a large amount of lime can be required to increase soil pH to neutrality.
Aluminum (Al) is an extremely problematic element of acid soils, and its behavior in the soil has a high interaction with CEC and DL theories. Liming is used to correct soil acidity since it promotes Al precipitation and increases pH and V%. However, as lime mobility in the soil profile is limited due to its low solubility, its action is restricted to the first centimeters of soil, especially in no-till systems (NTS), in which lime is applied on the soil surface. Nevertheless, the negative effect of soil acidity and aluminum toxicity has not been noticed in NTS. In an Hapludox of Ponta Grossa, state of Paraná, Brazil, Caires et al. (1999), working under a NTS, did not observe lime application effects on corn, wheat, and soybean yield, even under high levels of soil acidity (pH 4.5, V% of 32%, and m% of 18%). Alleoni et al. (2005a) studied an acid Hapludox (pH 4.4; V% of 36% and m% of 7%) from Rondonópolis, state of Mato Grosso, Brazil, and did not notice a meaningful effect of the lime application on soybean yield. Caires et al. (2005) evaluated the extent of the downward movement of surface-applied lime in a field experiment carried out from 1993 to 2003 in the Paraná State, Brazil, on a Typic Hapludox under NTS. The authors observed that surface-applied lime was effective in alleviating soil acidity below the point of placement. The effects of surface liming on all three acidity-related variables (pH, Al and basic cations) were significant for the 0-5 and 5-10 cm depths from one year onward, and also for the 10-20 cm depth from 2.5 years onward, remaining consistent for a period of up to ten years after liming. These results show that the negative acidity and Al toxicity effects are reduced in NTS. A possible explanation would lie on the presence of a greater amount of complex organic anions, which reduce the Al toxicity in soil solution (Salet, 1998).
Methods currently used in routine laboratory analyses to obtain bioavailable Al contents, for example, extraction with 1 mol L-1 KCl, usually do not detect the forms of this element which are toxic to plants. Aluminum can appear as a single soluble ion; as its hydrolyzed forms (AlOH2+, Al(OH)2+, Al(OH)3 and Al(OH)4-); complexed as sulphate (AlSO4+), fluoride (AlF63-), orthophosphate (AlH2PO42+), organic ligands (Al-organic) and it also can appear as polymers. The sum of all these aluminum chemical forms represents the total amount of aluminum in soil solution.
3.1.3 Quantitative Formulation of CEC
Mechanisms to describe quantitatively the cation exchange phenomenon have been studied over the years. Sometimes the efforts are concentrated to solve problems of soil management as in saline soils, for example, whereas other times the problem to be solved is the behavior of toxic elements, such as Al3+ or Mn2+. However, most times, emphasis is given to the understanding of the reactions and to an improvement on predicting the behavior of nutrients, such as Ca, Mg, K, NH4, etc. The knowledge of the chemistry of exchangeable cations can give important information about nutritional deficiencies and unbalances for the plants and also on the rates of toxic metal movements in the soil. For instance, the addition of Na-rich anthropogenic residues to tropical soils has stimulated the scientific community to study the role of Na in both, the soil solution and the exchange complex. Fonseca et al. (2005) evaluated methods to obtain the concentration of exchangeable and soluble cations in a greenhouse experiment with an Haplustox amended with a secondary-treated sewage effluent, collected from stabilization ponds at the wastewater treatment facility of Lins, São Paulo state, Brazil. The authors observed that, in closed environments of pots, the most suitable method for calculating the effective CEC was the cation exchange capacity calculated by removing cations with a barium chloride solution. The actual CEC should be measured using Mg adsorption, in order to prevent ionic strength from influencing electric charges. Silva et al. (2005) applied water treatment sludge to pots containing samples from a surface layer of an Hapludox and concluded that the CEC obtained by the sum of exchangeable cations in solution (K+, Ca2+, and Mg2+) was overestimated, since there was an excess of free cations, mainly Ca 2+, in the sludge.
Several models have been proposed to describe, characterize and predict the distribution of exchangeable ions in equilibrium between soluble and solid phases. Most of the models use the law of mass action. The ion partition was first described using the chemical equilibrium constant:

In these equations, the Na-K system is symmetrical or homovalent; the Ca-Na is non- symmetrical or heterovalent. X, the exchanger, is the particle which retains the exchangeable mono or divalent cations, and the parentheses mean activities of soluble and exchangeable cations. A constancy is found in varying compositions in the system phases only in the homovalent systems and in the ones in which the cations are very similar. Therefore, the system description, most of the times, involves an exchange selectivity coefficient. Although it is not a true constant, it is a parameter that may describe the exchange equilibrium in a range of different compositions of the system phases. The values of the selective coefficients vary with the composition of the phases in a given exchange reaction. In short, the equilibrium constants of the exchange reaction may be true constants under restricted system compositions or may become exchange selectivity coefficients (Ks). The parenthesis means cation activity only in the soluble phase, the brackets mean concentration or some function that models the concentrations or ion activities in the exchangeable phase, according to different approaches developed by various researchers:

3.1.3.1 Cation Exchange Equations
3.1.3.1.1 Kerr Equation
Kerr used for a symmetrical system (Ca-Mg) the law of mass action and ion concentrations in a solution in miliequivalents (meq) per volume of solution, whereas concentrations in meq per unit of mass were used for the ions adsorbed at solid phase, which he called "active masses" (Kerr, 1928). Although Kerr had not used activity in his studies, he assumed a proportionality between dissolved total concentrations and free Ca and Mg. The reaction and the constant are:

Since Kerr used a symmetrical system, the constancy of the coefficients was reasonable, he concluded that the exchangeable cations really formed a solid solution.
3.1.3.1.2 Vanselow Equation
Vanselow carried out studies using a thermodynamic approach, since he was not satisfied with Kerr's "active masses" definition (Vanselow, 1932). He used activities to represent the ions in solution and the mole fraction (MF) as the concentration of adsorbed cations for non-symmetrical systems, as follows:
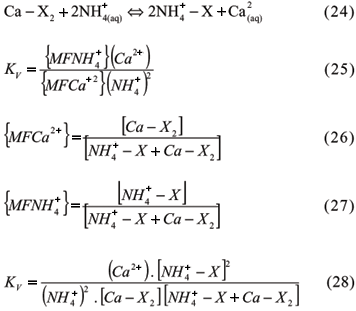
Mole fractions for the adsorbed cations are given in molar concentration (mol kg-1) of each one of them as related to total molar concentration adsorbed on the colloids.
Loyola Jr & Pavan (1989) studied the exchange reactions among Ca, Mg and K in five Oxisols and one Entisol with pH ranging from 4.0 to 4.4 under several ionic strengths (IS), but keeping the molecular ratio among the cations equal to 1. These authors obtained exchange selectivity coefficients for the three possible pairs of cations with the use of Vanselow equation and observed that the coefficients varied with the IS of the solution. The exchange sites presented greater attractive force in relation to Ca and similar forces of adsorption in relation to Mg and K.
3.1.3.1.4 Davis - Krishnamoorthy - Overstreet Equation
This equation describes the cation exchange based on thermodynamic statistics, in which different forms of neutralization of the colloidal surface charges by the cations and different charge configurations of colloidal surfaces had been considered (Davis, 1950). The final equation is similar to Vanselow equation, however with the introduction of a specific factor for bivalent ions in the unibivalent system. Krishnamoorthy & Overstreet (1950) tested this equation for many systems and colloids and concluded that the majority of the soils behave in such way so that the specific factor for bivalent ions is equal to 1.5. The final equation is sometimes called Davis equation and other times the Krishnamoorthy Overstreet equation. For a Ca-Na system, it can be described as:

3.1.3.1.5 Gapon Equation
Gapon (1933) developed an empirical expression to describe non-symmetrical unibivalent cation exchange, using concentrations instead of activities and the equation of mass action with chemically equivalent amounts for cations in solution and in exchangeable positions. The exchange system between Ca and Na presents the reaction:
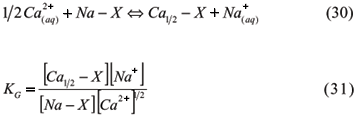
The soluble cations are expressed in mols L-1, while the exchangeable cations are expressed in meq g-1. Gapon's equation is widely used in saline soils studies because it presents a uniform coefficient for a large variation of exchangeable Na (0 to 40%), which is interesting for irrigated agriculture in many soils. For these soils, Gapon's constant is related to the percentage of saturation in the exchangeable sodium percentage (ESP) and Sodium Adsorption Ratio (SAR) (Kinjo & Marcos, 1982) as follows:

Silva et al. (1991) evaluated ESP, SAR, and KG values for alluvial soils with contrasting textures and noticed higher levels of KG at greater ionic strengths of the solution. Furthermore, for a specific IS, KG decreased with SAR increases. Gapon's equation can also be used in studies related to solute dynamics in soils. Ciprandi & Wietholter (1994) studied K selectivity coefficients along with several others cations in 28 soil samples and concluded that this coefficient, in addition to ion activities in solution and exchangeable cations, contents can be used to calculate the parameters of K diffusion and the soil buffer capacity by the element.
3.2 Specific Adsorption
Specific adsorption, also called chemisorption or ligand exchange, is the adsorption in which a structural bonding between the surface of the soil particles and the ion involved is formed. It is an important phenomenon that controls the bioavailability of some nutrients, toxic elements and heavy metals in the soil plant system.
3.2.1 Adsorption models
Adsorption is, by definition, the net accumulation of a chemical species at the interface between the solid phase and the solution (Sposito, 1989). Adsorption can be represented either by empirical or chemical models which describe a molecular adsorption using the theoretical concept of chemical equilibrium. The model is a representation in small scale or a simplified representation of reality. An ideal model is effective when it describes precisely the observations; it is comprehensive, when it can be applied to a large range of conditions without modifications; it is realistic, when it confirms to the accepted theories of behavior; and it is predictive, when it can be applied to the most different conditions (Barrow & Bowden, 1987).
Comparing the empirical and theoretical models, it is expected that the latter could satisfy most, if not all, of the above cited characteristics to give a closer description of the real adsorption phenomenon in the soil system. The main difference between the empirical and theoretical models is the lack of an electrostatic term in the former whereas its presence is mandatory in the latter. Among the main empirical models, the Constant Partition or Distribution Coefficient, the Langmuir and the Freundlich models will be discussed. Among the theoretical models, the Constant Capacitance, the Diffuse Layer, the Triple Layer and the Four Layers will be discussed.
3.2.1.1 Empirical models
Empirical models have been widely used in soil science and environmental studies related to metal and anion adsorption. These models do not take into consideration the electrostatic influence of the electrically charged surfaces in the solution, as well as the influence of changes in surface charges due to the composition of soil solution. The empirical models used in Soil Science are, in general, associated to the use of the so called adsorption isotherms. Adsorption isotherms are convenient ways to graphically represent the amount of an adsorbed compound, or adsorvate, in relation to the concentration in the equilibrium solution of this compound. As the name indicates, the temperature has to be constant, but if at the same time variables such as pH and ionic strength are also maintained constant, better results are expected.
A C-type (constant) isotherm is characterized by a steady slope from beginning to maximum adsorption, regardless of the degree of coverage on the surface. This type of isotherm may be the result of a constant partition of the adsorvate between the soil adsorbing surface and the soil solution or it can be a result of a proportional increase in the available adsorbent surface while the amount of adsorvate increases.
S-type isotherm (sigmoidal form) is characterized by a slight slope at low surface coverage, but increases with the higher concentration of adsorvate. This behavior seems to be related to the fact that, at low concentrations, the affinity of the adsorvate to soil particles is smaller as related to some component in soil solution. With an increase of the adsorvate concentration, the adsorption by the soil solution component decreases, and the adsorption by the soil particles begins to increase. At a certain point, the soil particles take over the adsorption and start to adsorb significantly the adsorvate.
The L-type isotherm (Langmuir) is the most commonly found in soil chemistry studies. The shape of this isotherm indicates that the adsorvate has a relatively high affinity for the surface at low charge coverage. However, as the surface coverage increases, the affinity of the adsorvate to the surfaces decreases.
To all of these isotherms, it is possible to relate mathematical models to describe them and to obtain parameters that may characterize the adsorption phenomenon.
3.2.1.1.1 Constant Partition Model
Most ions at very low concentrations and some non-polar organic or hydrophobic compounds have their adsorption characterized by the C-type or constant partition isotherm expressed by equation (33):

where q = adsorbed amount; Ceq = equilibrium concentration
This type of adsorption is modeled by a linear equation of slope Kd and intersept zero. More recently, this model has been used in many studies, mainly for heavy metals (Anderson & Christensen, 1988; Lee et al; 1996; Gao et al., 1997: McBride et al., 1997; Romkens & Salomons, 1998; Mc Bride et al., 1999; Gomes et al., 2001). Distribution or partition coefficients (Kd) have been the names used to designate the constant that mathematically describes this kind of adsorption.
Kd values are fundamentally important for the environment, because they reflect the soil solid surfaces affinity to chemical elements, such as heavy metals, whether or not they are essential to plants and animals. Kd values related to soil properties, pH, clay minerals, OM contents, nature of the contaminant, among others, led to the establishment of generic values for distribution coefficient by USEPA (United States Environmental Protection Agency, 1996). In the lack of established values for the Brazilian conditions, the São Paulo State uses generic standard values which may lead to inadequate evaluations due to climate and pedological differences between the temperate and tropical regions.
Soares (2004) evaluated heavy metal retention in 30 representative soils of the State of São Paulo, Brazil, based on a quantification of Kd and their relation with soil attributes, such as pH, CEC and clay, OM, and Fe, Al, and Mn oxides contents. The smallest variations in Kd value were recorded for Pb (121 to 7,020 L kg-1) and Ni (6 to 998 L kg-1), while larger variations were observed for Cd (7 to14,339 L kg-1), Co (2 to 34,473 L kg-1), Cr (1 to 21,267 L kg-1), and Zn (5 to 123,849 L kg-1). The following order of affinity was obtained: Pb >>> Cu >> Cd > Zn @ Ni @ Cr > Co. More than 55% of the variation in Kd for metallic cations of the groups IIB (Cd and Zn) and VIIIB (Co and Ni) were explained by pH. In the joint analysis, CEC and pH explained about 80% of the variation in Kd values for Cd, Co, and Ni. Regarding the distribution coefficients for Cu and Pb, pH and clay content accounted for about 63% of the variation. Kd values for Cr showed an inverse correlation with pH, especially in the joint analysis with clay contents, and 61% of the variation were explained by these variables.
3.2.1.1.2 Freundlich Model
Freundlich isotherm model is an empirical model formulated because the author assumed that the adsorption data followed a parabola, so that the equation of this curve was used to describe the phenomenon. Thus, the Freundlich model is:

where K and n are adjustable and positive parameters, with n values of 0<n<1. These parameters have no physical meaning, although Sposito (1980) presented n as measure of heterogeneity adsorption sites on the adsorvent surface.
Freundlich model constants can be determined by using either linear regression or adjusted by non-linear regression. A linear equation can be obtained by inserting logarithms at both sides:

The plot which relates log q to log Ceq represents a straight line with slope n and intercept log K. The adjustment of experimental results to the Freundlich model is validated through the significance of the coefficient of determination (R2). Several examples prove the capacity of this model to predict adsorption phenomena of various elements by weathered soils in the tropics, such as Alleoni et al. (1998), for B ; Alcântara & Camargo (2001), for Cr; Mesquita & Silva (2002), for Cu and Zn; Camargo et al. (1989) and Pombo et al. (1989) for Ni; Cunha et al. (1994) and Casagrande et al.(2004), for Zn; Pombo (1995) and Dias et al. (2001), for Cd, Jordão et al. (2000) and Silveira & Alleoni (2003), for Cu, and Alleoni et al. (2005b), for Cd and Cu.
Sorption and desorption coefficients for herbicides, taken as their partition coefficients normalized to the OC (Koc), can also be estimated by Freundlich's equation. Regitano et al. (2000) evaluated the sorption of imazaquin (2-[4,5dihydro-4-methyl-4-(1-methylethyl)-5-oxo-1H-imidazol-2-yl]-3-quinoline carboxylic acid) on seven surface and two subsurface samples of highly weathered soils and on other two contrasting soils. The authors observed that Freundlich isotherms were essentially linear (n ranged from 0.83 to 0.99) within the employed concentration range, suggesting that the hydrophobic partition of the soil organic matter is the major sorption mechanism. In addition, imazaquin sorption (Kf values) was positively correlated with OC (r = 0.91**), CEC (r = 0.78**), and SSA (r = 0.64*), but negatively correlated with soil-solution pH (r = -0.62*).
Rocha et al. (2002) studied imazaquin sorption and extraction in surface and subsurface samples of three soils at four different pH values. Sorption decreased, and aqueous extraction increased as the pH increased. Up to pH 5.8, sorption was higher in subsurface than in surface layers of the acric soils due to the positive balance of charges resulted from the high Fe and Al oxide and the low OC contents. This favored electrostatic interactions with the anionic molecules of imazaquin. For the subsurface samples of these highly weathered soils, for wich yo was positive and the OC content was low, it was not possible to predict sorption just by considering imazaquin speciation and its hydrophobic partition of the organic domains of the soil. Moreover, if Koc measured for the surface samples were assumed to represent the whole profile in predictive models for leaching potential, this would result in underestimation of the sorption potential in subsurface horizons, and consequently result in overestimation of the leaching potential.
3.2.1.1.3 Langmuir Model
Despite having been placed along with the empirical models, Langmuir's model is the only one that has its equation derived theoretically. However, as it was originally used to describe the adsorption of gas molecules to solid surfaces, its assumptions are not totally applicable to the soil system, and therefore, it is considered as an empirical model. The original assumptions of the Langmuir model are: (i) ions are adsorbed as a monolayer on the surface, and the maximum adsorption occurs when the surface is completely covered; (ii) adsorption takes place in specific positions, and there is only one molecule at each position or adsorption site; (iii) the surface is homogeneous, and the sites are identical; (iv) the adsorption energy is constant all over the surface and does not depend on the covered surface; (v) adsorbed species do not interact among themselves, and (vi) equilibrium is achieved. Langmuir's equation has the following formula:

in which b and K are adjustable parameters. The parameter b represents the value of q which is asymptotically approached as Ceq becomes larger. Usually, b is called the maximum adsorption capacity and is very common in adsorption studies in soils, and K is taken as a measure of the adsorption intensity. The product of K by the maximum adsorption b results in a value bK that represents the initial isotherm slope as Ceq approaches zero.
The constants b and K can be obtained by linear or non-linear regression. Using the linear regression analysis, the Langmuir equation is written in the form:

From equation (37), it can be seen that the plot of Ceq/q as a function of Ceq is a straight line with slope 1/b and intercept 1/bK. If the adjustment is satisfactory, which can be judged from the coefficient of determination (R2), the Langmuir model can be assumed to describe the adsorption process.
Langmuir's isotherm has been used to estimate P adsorption through the calculation of the maximum capacity of phosphate adsorption (Alvarez & Fonseca, 1990), as well as to relate phosphorus adsorption to different soil mineralogical components (Fontes & Weed, 1996). By using Langmuir's and Freundlich's models, among others, Fontes et al. (2000) studied the competitive adsorption of Cd, Cu, Zn, and Pb in tropical soils and reported the influence of these metals on the mobility and retention of Zn and Cd. In the most concentrated solutions, Zn and Cu adsorptions decreased. On the other hand, the competition did not affect Cu and Pb adsorptions, which implies in a more specific retention mechanism, involving covalent bonds between these elements and soil minerals. Araújo et al. (2002) employed the path coefficient analysis to evaluate both models and the ratio among the adsorption parameters obtained for some metals, such as Cd, Cu and Zn, and the characteristics of a series of tropical soils. Using the Langmuir model, Fontes & Gomes (2003) evaluated the competitive adsorption of Cr, Ni, Cu, Zn, Cd, and Pb in Brazilian representative soils (Oxisols, Ultisols and Alfisol) after OM removal and noticed that the metals with the most competitive effect were Cr, Cu and Pb. The most frequently obtained selectivity sequence was Cr = Pb >> Cu >>> Ni > Cd = Zn. Without OM removal, Ni was the least retained metal by soils, pointing out its low affinity in relation to this soil attributes.
Langmuir adsorption indices can be correlated to various soil properties. Alleoni & Camargo (2000) studied boron adsorption in five representative soils from the state of São Paulo, Brazil (Rhodic Hapludox, Arenic Paleudalf and three Typic Hapludoxes). High coefficients were found between B maximum adsorption and clay, and amorphous Al oxides contents and SSA (r = 0.79, 0.76 and 0.73, respectively, P < 0.01). Clay content, free Al oxide, and 0.01 mol L-1 CaCl2 extracted boron explained 93 % of the variation of the adsorbed boron. Highest values for maximum B adsorption were found for clayey soils, which were significantly correlated with contents of total, free and amorphous Fe and Al oxides, as well with physical attributes. Ninety four percent of the variation in the maximum adsorption could be related to the free iron content.
3.2.1.2 Models of Surface Complexation
Models of surface complexation are chemical models which treat ion adsorption as complexation reactions in the solid phase, similar to the formation of a complex in solution. These models are tools to identify chemical reactions and their equilibrium constants. Assuming the validity of retention mechanisms and equilibrium constants, models of surface complexation can be used to predict the distribution of a substance between the adsorbed and the hydrous phases. These models also consider the adsorvate charges and the charges of the adsorvent surface so that they represent a more significant advance over the empirical models (Goldberg, 1995).
The majority of colloidal particles carry significant electric charge at their surface, then exhibiting electric potentials which extend into soil solution. Ions present in the solution interact with these charges, and those of same charge are repelled, while the opposite are attracted. Therefore, electrostatic potentials associated to charge surfaces can have a significant influence on the adsorption of ions. The influence of electric charged surfaces in the adsorptive behavior of charged species is merged in the electrostatic adsorption models by adding some terms that modify the activity of adsorbed ions that approach the charged surface. The electric work required to penetrate the area of electrostatic potentials (ys), that appears while ions reach large distances, is also considered.
There are several models to describe the surface complexation. In general, these models vary in the way they conceive charge distribution and electric potential and the location of adsorbed species in the interface solid-solution. In spite of some differences, there are several assumptions applied to all models involved in the solid interface-solution: (i) all mineral surfaces present in aqueous environment or suspension have functional groups or surface sites, and (ii) total concentration for each type of surface site can be defined (Essington, 2004).
3.2.1.2.1 Constant Capacitance Model (CCM)
Developed for oxide surfaces at the interface with aqueous solutions by Paul W. Schindler and Werner Stumm, along with several collaborators (Schindler & Gamsjager, 1972; Hohl & Stumm, 1976; Schindler et al., 1976; Stumm et al., 1976; 1980), this model is based on the following assumptions; (i) all surface complexes are considered inner-sphere complexes, and adsorption occurs by ligand exchanges; (ii) no surface complex are formed with the electrolyte ions; (iii) a plan of charges represents the surface; (iv) surface density charge (s) is proportional to (yo) at the particle surface, or s = C.yo, where s is expressed in C m-2 (coulombs per square meter); C is the capacitance in F m-2 (Farad per square meter,) and yo is in V (volts).
For S = oxide surface, SOH = protonated or deprotonated hydroxide surface, M = Metals, L = Ligands, and m and l = valencies, the chemical reaction based on Hohl et al. (1980) are:
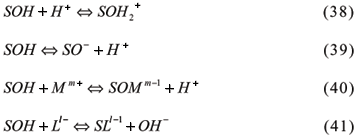
Bidentated compounds may be formed, mainly with transition metals and anions of poliprotic acids, and some other reactions are important such as:

Each reaction can be described by an intrinsic equilibrium constant (Hohl et al., 1980) in which the brackets represent the concentration in mol L-1 or molar (M). Intrinsic constants take into account the electrostatic parameter, i.e., the potential, since other values from the exponential component are constant.

Two additional equations (mass balance and charge balance for the surface functional groups) are necessary to solve the equilibrium problem. (Sigg & Stumm, 1981; Goldberg, 1995).
CCM is similar to Helmholtz's double layer model, since both locate the adsorbed ions close to particle surface. This model has been used to describe boron adsorption on oxides (Goldberg & Glaubig, 1985) and on silicate clay minerals (Goldberg & Glaubig, 1986b), which constitutes important adsorption surfaces for this element. Oxides and broken edges of clay minerals are variable charge materials important as B adsorption sites (Goldberg, 1993). Soil corresponds to a mixture of organic and inorganic materials, but even in such a complex system, CCM fitted well (Goldberg & Glaubig, 1986a). Either for pure composts or for soils, values of intrinsic constants were optimized with the complexation constants of anions in the surface. Goldberg & Glaubig (1986a) used a mean value of many conditional intrinsic surface constants in the CCM and observed that the model predicted well the B adsorption on many soils from California, USA.
In order to evaluate CCM adequacy to Mo adsorption in 36 soils, Goldberg et al. (2002) employed a regression model to obtain the surface constants from CEC, Fe oxide, organic and inorganic carbon, and found that the model was able to predict the adsorption. More recently, Goldberg at al. (2005) applied the CCM to arsenate (As(V)) adsorption on 49 soils selected by soil property variation. The high affinity of arsenic for oxide surfaces is affected by several biogeochemical factors such as soil texture, OM content, nature and constituents of minerals, pH, redox potential, and competing ions. The activity of arsenic in soil solution is most commonly controlled by surface complexation reactions on oxides/hydroxides of Al, Mn, and especially Fe, which makes its availability highly dependent on soil attributes (Gonzaga et al., 2006). Goldberg at al. (2005) found that CCM was able to fit arsenate adsorption on all soils by optimizing either three monodentate or two bidentate As(V) surface complexation constants. A general regression model was developed for predicting soil As(V) surface complexation constants from CEC, inorganic and organic carbon content, Fe oxide content, and SSA. According to the authors, the incorporation of regression equations in to chemical models allows simulation procedures of speciation and transition of chemical elements in several conditions of soil and environmental management, without any specific adsorption studies.
3.2.1.2.2 Diffuse Layer Model (DLM)
The diffuse Layer Model was initially proposed by Stumm et al. (1970) and Huang & Stumm (1973) for a soil oxide-solution interface. Dzomback & Morel (1990) made an improvement called generalized double layer model (GDLM). The first two assumptions from the CCM were maintained in this model, and two other were included: (i) two planes of charge represent the surface, and (ii) the ratio between charges and surface potentials are (Goldberg, 1995):
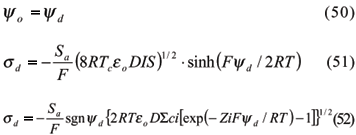
in which eo = vacuum permittivity; D = dielectric constant of water; IS = ionic strength; sgnyd= 1 if yd > 0, or yd = -1 if yd < 0; ci = concentration of ionic species I in solution; Zi = charge or valency of the ionic species I in solution.
Equation (51) is equivalent to Gouy-Chapman's for the double-layer charges and it is applied to indifferent symmetric electrolytes. On the other hand, equation (52) is more complex and is applied to calculate surface charge for non-symmetric electrolytes. Equations for protonation, deprotonation and/or dissociation, and metal adsorption are the same of CCM. However, bidentate complexes are not defined in the GDLM, and the complexation ligand reactions are (Dzomback & Morel, 1990; Goldberg, 1995):

Intrinsic equilibrium constants of several reactions are the same as used in CCM, whereas the constant to describe complexation reactions of ligands are (Dzomback & Morel, 1990; Goldberg, 1995):

Mass balance of functional surface groups and charge balance are also important for the final solution. Equations may be either solved by manual calculations, as described in Dzomback & Morel (1990) or by computer software (Goldberg, 1995).
Spathariotis & Kallianou (2001) studied the adsorption of Cu, Zn, and Cd, as a function of pH, ionic strength of the solution, and metal concentration. Adsorption occurred as Cu > Zn > Cd, over all situations. CCM simulated the formation of mono and bidentate complexes, according to the used procedures to obtain a specific surface. When they used the Brunauer-Emmett-Teller (BET) method, which utilizes nitrogen gas, the formation of bidentate complexes was observed, and it was more convenient, since spectroscopic methods had detected the presence of such compounds. The authors also observed that DLM had an inferior adjustment than CCM for all situations.
3.2.1.2.3 Triple Layer Model
The Triple-Layer Model (TLM) was developed as a model of bonding sites for oxide interfaces with soil solution (Yates et al., 1974), and later its concept was expanded by Davis and his collaborators (Davis et al., 1978; Davis & Leckie, 1978; 1980). The original model, assumed that surface groups of soil particles formed inner-sphere complexes only with protons and hydroxyl ions, while metals and ligands formed outer-sphere complexes with the particle surface. The model was modified afterwards (Blesa et al., 1984; Hayes & Leckie, 1986; 1987; Hayes et al., 1988) in order to include inner-sphere complexes. The modified version of the TLM allows for the adsorption of metals and ligands in the inner-sphere plan, forming inner-sphere complexes. It also allows for the formation of oter-sphere complexes.
Assumptions for the TLM are (Goldberg, 1995): (i) protons and hydroxyl ions form inner-sphere surface complexes; (ii) ion adsorption reactions form either inner or outer-sphere surface complexes (iii) ions from background electrolyte form outer-sphere surface complexes; (iv) three planes of charge represent the surface; and (v) relationships betweens charges and surface potential are:
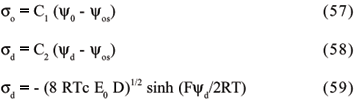
where s has units of C m-2, eo is the permittivity of vacuum, D is the dielectric constant of water, and c is the concentration of a 1:1 background electrolyte.
Equations for the complexation reactions which form inner-sphere complexes are described for CCM, whereas the equations to form outer-sphere complexes are:

In these equations, C+ and A- correspond to cations and anions from the background electrolyte, and the outer-sphere complexes are represented by a larger dash. Intrinsic equilibrium constants for protonation, deprotonation and inner-sphere complexation of metals and ligands are identical to constants already defined for the CCM. Equilibrium constants for the outer-sphere complexes reactions are defined as:
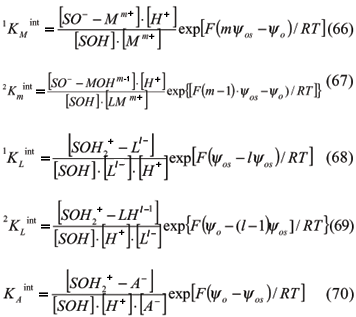
The TLM describes successfully the adsorption of sulphate on aluminum and titanium oxides, among others (Charlet et al., 1993). According to these authors, the sulphate bonding to soil colloidal surfaces is predominantly electrostatic, but long-term experiments are fundamental to establish if covalent bondings may occur, mainly when anion concentrations are high. This is an important observation when gypsum is used as a soil conditioner, because the sulphate may be involved in the process of charge generation, instead of simply acting as an indifferent ion.
3.2.1.2.4 Four-layer model (FLM)
The FLM model simulates the adsorption in terms of pH, considering electric potential variation of soil colloids and amount of ions in solution (Casagrande & Camargo, 1997) In this model, electrostatic potentials in the plane where ions are adsorbed (ya) can not be measured, since the model does not take into account the equilibrium constant between the adsorbed species and soil colloids, and these constants are required when the potential is calculated. So, it is necessary to assume an initial value for the electric potential, which is a chosen and adjustable parameter.
The neutral surface of FLM is represented by OH-S-OH2 in which the surface electric potential decreases due to an increase of soil acidity (equation 71), but increases when pH becomes higher (equation 72) (Bowden et al., 1977).

In this model, variations in pH, ionic strength, and concentration and affinity of adsorbed elements are taken into consideration. Therefore, it can be greatly applied on soils and minerals (Barrow, 1987). The reaction between an ion and a soil charged surface includes a component that reflects a chemical attraction between both. The activity of an ion located close to the surface (ais) can be related to its activity in solution (ai), equation (73):

K is the reaction constant, which is related to the ion and to the surface, Zi is the ion valency, øa is the electrostatic potential, F is Faraday constant, R is the gases constant, and T is the absolute temperature (Bowden et al.,1977). The term ai can be replaced by a g c, where a is the proportion of the ion, g is the coefficient of activity and c is the ion concentration in solution. The activity ais is equal to the quantity of occupied sites in relation to available sites, that is q/(1-q), in which q is the proportion of occupied sites (Bowden et al., 1977). Then:

By using FLM, we can obtain adjustable parameters, the ion bonding constant to colloidal surfaces K, and the soil surface electric potential. These two parameters can be used to predict the adsorption of an ion under a wide range of pH and ion concentrations added to the soil. Casagrande & Camargo (1997) studied the phosphate adsorption in acric soils and found out that this model was far beyond a simple curve adjustment, since it allowed the interpretation of pH variations and phosphate concentration in the equilibrium solution, although the electrostatic potential values were not realistic. Casagrande et al. (2003) evaluated the adequacy of the model to phosphate and sulphate adsorption in surface and subsurface samples of a Rhodic Acrudox, an Anionic Acrudox, and a Rhodic Hapludalf. They observed that surface electric potentials estimated by the FLM were not realistic, though the model performed well including pH, phosphate and sulphate concentrations effects on the potential variation, besides being efficient in simulating anion adsorption.
4. Ionic speciation x availability of nutrients.
Fertile soils are able to supply plants with essential elements, which are vital for normal growth and reproduction. Among the 17 essential elements (C, H, O, N, P, S, Cu, Ni, B, Mo, Cl), 14 are obtained from the soil, preferably. Carbon, hydrogen and oxygen are supplied by CO2 and H2O, abundant elements in both atmosphere and hydrosphere. Except N, which is present in a large amount in the atmosphere (79%), the natural source of all other elements is the weathering of primary minerals in the parent material. The power of nutrient supplying of a given soil is the measure of its fertility (White, 1997).
Essential elements can be grouped in several ways but in order to be discussed along with the soil electrochemical properties they should be grouped as: (i) Ca, Mg, K, Fe, Mn, Zn, Cu, and N which are the ones that form cations and (ii) N, P, S, B, Mo, Cl which form anions. Going one step further, a geochemical concept based on ionic potential (IP) of the chemical elements can be used, which is obtained from the ratio between the valency and the ionic radius. By using this concept, cations can be separated in the following groups: (i) Ca, Mg, K, Fe2+, Mn2+, Zn, Cu, Ni (IP < 3.0) the ones that simply solvate in an aqueous solution; (ii) Fe, Mn3+, Mn4+, Al (3.0 < IP < 9.5), the ones that hydrolyze extensively at pH 7; and (iii) N, P, S, B, Mo (IP > 9.5) the ones that are capable of forming oxyanions (Sposito, 1989; McBride, 1994).
Still going a little further, it is possible to separate the metals which tend to form electrovalent bonding from those of more electrostatic bonding by using the Misono Softness index (Sposito, 1989). This parameter is defined as a function of the ionic radius, valency and ionization potential of the chemical element and allows the separation of metals less inclined to form covalent bonding, such as Ca, Mg and K, from Fe, Mn, Zn, Cu, and Ni, which act in the opposite way, i.e., tend to form strong covalent bondings.
In this context, the final grouping of the chemical elements to better relate them to the soil electrochemical properties would be:
-
electrostatic cationic macronutrients: Ca, Mg, K, N-NH
4
-
covalent micronutrients: Zn, Cu, Ni
-
micronutrients of easy precipitation: Fe, Mn
-
electrostatic anionic macronutrient: N-NO
3
-
covalent anionic macronutrients: P, S
-
electrostatic anionic micronutrient: Cl
-
covalent anionic micronutrients: B, Mo
Either the metallic or the non-metallic elements can exhibit different oxidation numbers and may appear as free ions or ionic pairs, as complexes or chelates, or as crystalline or amorphous compounds. The description and characterization of chemical forms of elements in solution, i.e., chemical speciation, has a huge influence on their bioavailability. As first approach, plant behavior is correlated to concentrations or activities of free metals or non-complexed cations in soil solution (Parker et al., 1995).
Absorption of free cations by plant roots is a reasonable expectation, considering that plant cells have negative transmembranic potentials originating from internal negative potentials that would favor the absorption processes energetically (Marschner, 1986). On the other hand, complexed cations have their positive charges reduced or cancelled, and, therefore, become larger and less reactive. Transmembranic potential of cells hampers both the transport and absorption of anions, and any reduction on ionic charge decreases the barrier of the potential energy (Parker et al., 1995). Some examples include a greater absorption of H2PO4- as related to HPO42- (Hendrix, 1967) and a greater absorption of H3BO3, as compared to B(OH)4 (Oertli & Grgurevic, 1975).
5. Final remarks
The relationship between the soil electrochemical properties and the availability of nutrients, toxic elements and heavy metals in soils has to be considered under different approaches. First, a better understanding of the role of the double layer of charges and CEC on nutrient cation availability for highly weathered soils is required, as well as a solid comprehension of adsorption models with emphasis on surface complexation models, in order to improve the knowledge regarding the behavior of anions in soils.
Furthermore, more studies have to be conducted to generate results that enable the use of chemical speciation concepts and calculation of several constants presented in surface complexation models, especially for highly weathered soils from the humid tropics. Last of all, there should have a continuing development and use of computer softwares that have already incorporated the concepts of chemical speciation and adsorption models in the study of nutrients, toxic elements and heavy metals availability in soil-plant system.
6. REFERENCES
Received August 01, 2006
Accepted November 15, 2006
- ABREU, C.A.; RAIJ, B. van; ABREU, M.F.; GONZÁLEZ, A.P. Routine soil testing to monitor heavy metals and boron. Scientia Agricola, v.62, p.564-571, 2005.
- ALCÂNTARA, M.A.K.; CAMARGO, O.A. Isotermas de adsorção de Freundlich para crômio (III) em latossolos. Scientia Agricola, v.58, p.567-572, 2001.
- ALLEONI, L.R.F.; CAMARGO, O.A. Pontos de efeito salino nulo de latossolos ácricos. Revista Brasileira de Ciência do Solo, v.18, p.175-180, 1994a.
- ALLEONI, L.R.F.; CAMARGO, O.A. Modelos de dupla camada difusa de Gouy-Chapman e Stern aplicados a latossolos ácricos paulistas. Scientia Agricola, v.51, p.315-320, 1994b.
- ALLEONI, L.R.F.; CAMARGO, O.A. Solos ácricos: atributos e manejo. Boletim Informativo da Sociedade Brasileira de Ciência do Solo, v.20, p.9-17, 1995.
- ALLEONI, L.R.F.; CAMARGO, O.A. Boron adsorption in soils from the State of São Paulo, Brazil. Pesquisa Agropecuária Brasileira, v.53, p.413-422, 2000.
- ALLEONI, L.R.F.; CAMARGO, O.A.; CASAGRANDE, J.C. Isotermas de Langmuir e de Freundlich na descrição da adsorção de boro em solos altamente intemperizados. Scientia Agricola, v.55, p.379-387, 1998.
- ALLEONI, L.R.F.; CAMBRI, M.A.; CAIRES, E.F. Atributos químicos de um Latossolo de cerrado sob plantio direto, de acordo com doses e formas de aplicação de calcário. Revista Brasileira de Ciência do Solo, v.29, p.923-934, 2005a.
- ALLEONI, L.R.F.; IGLESIAS, C.S.M.; MELLO, S.C.; CAMARGO, O.A.; CASAGRANDE, J.C.; LAVORENTI, N.A. Atributos do solo relacionados à adsorção de cádmio e de cobre em solos tropicais. Acta Scientiarum, v.27, p.729-737, 2005b.
- ALVAREZ, V.H.; FONSECA, D.M. Definição de doses de fósforo para a determinação da capacidade máxima de adsorção de fosfatos e para ensaios de casa de vegetação. Revista Brasileira de Ciência do Solo, v.14, p.49-55, 1990.
- ANDERSON, P.R., CHRISTENSEN, T.H. Distribution coefficients of Cd, Co, Ni and Zn in soils. Journal of Soil Science, v.39, p.15-22, 1988.
- ARAÚJO, W.S.; AMARAL SOBRINHO, N.M.B.; MAZUR, N.; GOMES, P.C. Relação entre adsorção de metais pesados e atributos químicos e físicos de classes de solo do Brasil. Revista Brasileira de Ciência do Solo, v.26, p.17-27, 2002.
- BLESA, M.A.; MAROTO, A.J.G.; REGAZZONI, A.E. Boric acid adsorption on magnetite and zirconium dioxide. Journal of Colloid and Interface Science, v.99, p.32-40, 1984.
- BARROW, N.J. Reaction of anions and cations with variable-charge soils. Advances in Agronomy, v.38, p.183-230, 1985.
- BARROW N. J. ; BOWDEN J. W. A comparison of models for describing the adsorption of anions on a variable charge mineral surface. Journal of Colloid and interface science, v.119, p.236-250, 1987.
- BOWDEN, J.W.; POSNER, A.M.; QUIRK, J.P. Ionic adsorption on variable charge mineral surfaces. Theoretical charge development and titration curves. Australian Journal of Soil Research, v.15, p.121-136, 1977.
- CAHN, M.D.; BOUDIN, D.R.; CRAVO, M.S. Nitrate sorption in the profile of an acid soil. Plant and Soil, v.143, p.179-183, 1992.
- CAIRES, E.F.; ALLEONI, L.R.F.; CAMBRI, M.A.; BARTH, G. Surface application of lime for crop grain production under a no-till system. Agronomy Journal, v.97, p.791-798, 2005.
- CAIRES, E.F.; FONSECA, A.F.; MENDES, J.; CHUEIRI, W.A.; MADRUGA, E.F. Produção de milho, trigo e soja em função das alterações das características químicas do solo pela aplicação de calcário e gesso na superfície, em sistema de plantio direto. Revista Brasileira de Ciência do Solo, v.23, p.315-327, 1999.
- CAMARGO, O.A.; RAIJ, B. van. Movimento de gesso em amostras de latossolos com diferentes propriedades eletroquímicas. Revista Brasileira de Ciência do Solo, v.13, p.275-80, 1989.
- CAMARGO, O.A.; ROVERS, H.; VALADARES, J.M.A.S. Adsorção de níquel em Latossolos paulistas. Revista Brasileira de Ciência do Solo, v.13, p.125-129, 1989.
- CAMARGO, O.A.; ALLEONI, L.R.F.; CASAGRANDE, J.C. Reações dos micronutrientes e elementos tóxicos. In: FERREIRA, M.E.; CRUZ, M.C.P.; RAIJ, B. van; ABREU, C.A. (Ed.). Micronutrientes e elementos tóxicos na agricultura Jaboticabal: CNPq; FAPESP; POTAFOS, 2001. cap.5, p.89-124.
- CASAGRANDE, J.C.; CAMARGO, O.A. Adsorção de fosfato em solo com caráter ácrico avaliada por um modelo de complexação de superfície. Revista Brasileira de Ciência do Solo, v.21, p.353-360, 1997.
- CASAGRANDE, J.C.; ALLEONI, L.R.F.; CAMARGO, O.A.; BORGES, M. Adsorção de fosfato e sulfato em solos com cargas elétricas variáveis. Revista Brasileira de Ciência do Solo, v.25, p.51-59, 2003.
- CASAGRANDE, J.C.; ALLEONI, L.R.F; CAMARGO, O.A.; ARNONE, A.D. Effects of pH and ionic strength on zinc sorption by a variable charge soil. Communications in Soil Science and Plant Analysis, v.35, p.2087-2095, 2004.
- CHARLET, L.; DISE, N.; STUMM, W. Sulfate adsorption on a variable charge soil and on reference minerals. Agriculture, Ecosystems & Environment, v.47, p.87-102, 1993.
- CIPRANDI, M.A.O.; WIETHÖLTER, S. Cation selectivity coefficients and diffusion parameters of potassium in soils of the state of Rio Grande do Sul. Revista Brasileira de Ciência do Solo, v.18, p.395-405, 1994.
- CUNHA, R.C.A.; CAMARGO, O.A.; KINJO, T. Aplicação de três isotermas na adsorção de zinco em Oxissolos, Alfissolos e Ultissolos. Revista Brasileira de Ciência do Solo, v.18, p.15-20, 1994.
- DAVIS, J.A.; JAMES, R.O.; LECKIE, J.O. Surface ionization and complexation at the oxide/water interface. I. Computation of electrical double layer properties in simple electrolytes. Journal of Colloid and Interface Science, v.63, p.480-499, 1978.
- DAVIS, J.A.; LECKIE, J.O. Surface ionization and complexation at the oxide/water interface. II. Surface properties of amorphous iron oxyhydroxide and adsorption of metal ions. Journal of Colloid and Interface Science, v.67, p.90-107, 1978.
- DAVIS, J.A.; LECKIE, J.O. Surface ionization and complexation at the oxide/water interface. III. Adsorption of anions. Journal of Colloid and Interface Science, v.74, p.32-34, 1980.
- DIAS, N.M.P.; ALLEONI, L.R.F.; CASAGRANDE, J.C.; CAMARGO, O.A. Isotermas de adsorção de cádmio em Latossolos ácricos. Revista Brasileira de Engenharia Agrícola e Ambiental, v.5, p.229-234, 2001.
- DZOMBACK, D.A.; MOREL, F.M.M. Surface complexation modeling Hydrous ferric oxide. New York: John Wiley & Sons, 1990.
- ESSINGTON, M.E. Soil and water chemistry: an integrative approach Boca Raton: CRC Press, 2004. 534p.
- FONSECA, A.F.; ALLEONI, L.R.F.; MELFI, A.J.; MONTES, C.R. Cation exchange capacity of an Oxisol amended with an effluent from domestic sewage treatment. Scientia Agricola, v.62, p.552-558, 2005.
- FONTES, M.P.F.; WEED, S.B. Iron oxides in selected Brazilian Oxisols: I. Mineralogy. Soil Science Society of America Journal, v.55, p.1143-1149, 1991.
- FONTES, M.P.F.; WEED, S.B. Phosphate adsorption by clays from Brazilian Oxisols: relationship with specific area and mineralogy. Geoderma, v.72, p.37-51, 1996.
- FONTES, M.P.F.; GOMES, P.C. Simultaneous competitive adsorption of heavy metals by the mineral matrix of tropical soils. Applied Geochemistry, v.18, p.795-804, 2003.
- FONTES, M.P.F.; CAMARGO, O.A.; SPOSITO, G. Eletroquímica das partículas coloidais e sua relação com a mineralogia de solos altamente intemperizados. Scientia Agricola, v.58, p.627-646, 2001.
- FONTES, M.P.F.; MATOS, A.T.; COSTA, L.M.; NEVES, J.C.L. Competitive adsorption of zinc, cadmium, copper and lead in three highly-weathered Brazilian soils. Communications in Soil Science and Plant Analysis, v.31, p.2939-2958, 2000.
- FORD, R.G.; SCHEINOST, A.C.; SPARKS, D.L. Frontiers in metal sorption/precipitation mechanisms on soil mineral surfaces. Advances in Agronomy, v.74, p.41-62, 2001.
- GAO S.; WALKER, W.J.; DAHLGREN, R.A.; BOLD, J. Simultaneous sorption of Cd, Cu, Ni, Zn, Pb and Cr on soils treated with sewage sludge supernatant. Water Air and Soil Pollution, v.93, p.331-345, 1997.
- GOLDBERG, S. Chemistry and mineralogy of boron in soils. In: GUPTA, U. (Ed.). Boron and its role in crop production Boca Raton: CRC Press, 1993. p.3-44.
- GOLDBERG, S. Adsorption models incorporated into chemical equilibrium models. In: LEOPPERT, R.H.; SCHWAB, A.P.; GOLDBERG, S. (Ed.). Chemical equilibrium and reaction models Madison: SSSA, 1995. (Special Publication, 42).
- GOLDBERG, S.; GLAUBIG, R.A. Boron adsorption on aluminum and iron oxide minerals. Soil Science Society of America Journal, v.49, p.1374-1379, 1985.
- GOLDBERG, S.; GLAUBIG, R.A. Boron adsorption on California soils. Soil Science Society of America Journal, v.50, p.1173-1176, 1986a.
- GOLDBERG, S.; GLAUBIG, R.A. Boron adsorption and silicon release by the clay minerals kaolinite, montmorillonite, and illite. Soil Science Society of America Journal, v.50, p.1442-1448, 1986b.
- GOLDBERG, S.; LESCH, S.M.; SUAREZ, D.L. Predicting molybdenum adsorption by soils using soil chemical parameters in the constant capacitance model. Soil Science Society of America Journal, v.66, p.1836-1842, 2002.
- GOLDBERG, S.; LESCH, S.M.; SUAREZ, D.L.; BASTA, N.T. Predicting arsenate adsorption by soils using soil chemical parameters in the Constant Capacitance Model. Soil Science Society of America Journal, v.69, p.1389-1398, 2005.
- GOMES, P.C.; FONTES, M.P.F.; SILVA, A.G.; MENDONÇA, E.S.; NETTO A.R. Selectivity sequence and competitive adsorption of heavy metals by Brazilian soils. Soil Science Society of America Journal, v.65, p.1115-1121, 2001.
- GONZAGA, M.I.S.; SANTOS, J.A.G.; MA, L.Q. Arsenic phytoextraction and hyperaccumulation by fern species. Scientia Agricola, v.63, p.90-101, 2006.
- HARTEMINK, A.E.; BURESH, R.J.; JAMA, B.; JANSSEN, B.H. Soil nitrate and water dynamics in Sesbania fallows, weed fallows ad maize. Soil Science Society of America Journal, v.60, p.568-574, 1996.
- HARTER, R.D.; NAIDU, R. An assessment of environmental and solution parameter impact on trace-metal sorption by soils. Soil Science Society of America Journal, v.65, p.597-612, 2001.
- HAVLIN, J.H.; BEATON, J.D.; TISDALE, S.L.; NELSON, W.L. Soil fertility and fertilizers: an introduction to nutrient management. Upper Saddle River: Prentice Hall, 1999, 499p.
- HAYES, K.F.; LECKIE, J.O. Mechanism of lead ion adsorption at the goethite-water interface. In: DAVIS, J.A.; HAYES, K.F. Geochemical processes at mineral surfaces Washington: American Chemical Society, 1986. p.114-141. (Symposium Series, 323).
- HAYES, K.F.; LECKIE, J.O. Modeling ionic strength effects on cation adsorption at hydrous oxide/so lution interfaces. Journal of Colloid and Interface Science v.115, p.564-572, 1987.
- HAYES, K.F.; PAPELIS, C.; LECKIE, J.O. Modeling ionic strength effects on cation adsorption at the hydrous oxide/solution interface. Journal of Colloid Interface Science, v.125, p.717-726, 1988.
- HENDRIX, J.E. The effect of pH on the uptake and accumulation of phosphate and sulfate ions by beans plants. American Journal of Botany, v.54, p.560-564, 1967.
- HERBILLON, A.J. Introduction to the surface charge properties of iron oxides and oxidic soils. In: STUCKI, J.W.; GOODMAN, B.A.; SCHWERTANN, U. (Ed.) Iron in soils and clay minerals Boston: Reidel, 1988. p.251-264. (NATO Advanced Studies Institute, Série C, 217).
- HOHL, H.; STUMM, W. Interaction of Pb2+ with hydrous g-Al2O3 Journal of Colloid and Interface Science, v.55, p.281-288, 1976.
- HOHL, H.; SIGG, L.; STUMM, W. Characterization of surface chemical properties of oxides in natural waters. Advances in Chemistry Series, v.189, p.1-31, 1980.
- HUANG, C.P.; STUMM, W. Specific adsorption of cations on hydrous g-Al2O3 Journal of Colloid Interface Science, v.43, p.409-420, 1973.
- HUNTER, R.J. Introduction to modern colloid science New York: Oxford University Press, 1993. 338p.
- JORDÃO, C.P.; ALVES, N.M.; PEREIRA, J.L.; BELLATO, C.R. Adsorção de íons Cu2+ em Latossolo Vermelho-Amarelo húmico. Química Nova, v.23, p.5-11, 2000.
- KINJO, T.; MARCOS, Z.Z. Adsorção de sódio em solos do estado de São Paulo. II. Equilíbrio de troca catiônica entre sódio e cálcio. Revista Brasileira de Ciência do Solo, v.6, p.78-82, 1982.
- LEE, S.Z.; ALLEN, H.E.; HUANG, C.P.; SPARKS, D.L.; SANDERS, P.F.; PEIJNENBURG, W.J.G.M. Predicting soil-water partition coefficients for cadmium. Environmental Science and Technology, v.30, p.3418-3424, 1996.
- LOYOLA Jr., E.; PAVAN, M.A. Seletividade de troca de cátions em solos ácidos. Revista Brasileira de Ciência do Solo, v.13, p.131-138, 1989.
- MARSCHNER, H. Mineral nutrition of higher plants New York: Academic Press, 1986.
- McBRIDE, M.B. Environmental chemistry of soils New York: Oxford University Press. 1994. 406p.
- McBRIDE, M.B.; RICHARDS, B.K.; STEENHUIS, T.; SPIERS, G. Long term leaching of trace elements in a heavily sludge-amended silty clay loam soil. Soil Science, v.164, p.613-623, 1999.
- McBRIDE, M.B.; RICHARDS, B.K.; STEENHUIS, T.; RUSSO, J.J.; SAUVÉ, S. Mobility and solubility of toxic metals and nutrients in soil fifteen years after sludge application. Soil Science, v.162, p.487-500, 1997.
- MEKARU, T.; UEHARA, G. Anion adsorption in ferruginous tropical soil. Soil Science Society of America Proceedings, v.36, p.296-300, 1972.
- MESQUITA, M.E.; SILVA, J.M.V. Preliminary study of pH effect in application of Langmuir and Freundlich isotherms to Cu-Zn competitive adsorption. Geoderma, v.106, p.219-234, 2002.
- OERTLI, J.J.; GRGUREVIC, E. Effect of pH on the absorption of boron by excised barley roots. Agronomy Journal, v.67, p.278-280, 1975.
- OLPHEN, H. van. An introduction to clay colloid chemistry New York: Jonh Wiley & Sons, 1977. 318p.
- PARFITT, R.L.; ATKINSON, R.J.; SMART, R.St.C. The mechanisms of phosphate fixation by iron oxides. Soil Science Society of America Journal, v.39, p.837-841, 1975.
- PARKER, R.D.; CHANEY, R.L.; NORVELL, W.A. Chemical equilibrium models: Applications to plant nutrition research. In: LEOPPERT, R.H.; SCHWAB, A.P.; GOLDBERG, S. (Ed.). Chemical equilibrium and reaction models Madison: SSSA, 1995. (Special Publication, 42).
- PARKS, G.A.; BRUYN, P.L. The zero point of charge of oxides. Journal of Physical Science, v.66, p.967-973, 1962.
- POMBO, L.C.A. Sorção de cádmio em solos do estado do Rio Grande do Sul. Revista Brasileira de Ciência do Solo, v.19, p.19-24, 1995.
- POMBO, L.C.A.; SALGADO, V.; VOLKWEISS, S.; KLAMT, E. Adsorção de níquel por dois solos: Terra Bruna estruturada similar e Podozólico Vermelho-Amarelo. Pesquisa Agropecuária Brasileira, v.24, p.593-598, 1989.
- REGITANO, J.B.; ALLEONI, L.R.F.; VIDAL-TORRADO, P.; CASAGRANDE, J.C.; TORNISIELO, V.L. Imazaquin sorption in highly weathered tropical soils. Journal of Environmental Quality, v.29, p.894-900, 2000.
- RAJAN, S.S.S. Changes in net surface charge of hydrous alumina with phosphate adsorption. Nature, v.262, p.45-46, 1976.
- ROCHA, W.S.D.; REGITANO, J.B.; ALLEONI, L.R.F.; TORNISIELO, V.L. Sorption of imazaquin in soils with positive balance of charges. Chemosphere, v.49, p.263-270, 2002.
- RÖMKENS, P.F.A.M.; SALOMONS, W. Cd, Cu and Zn solubility in arable and forest soils: consequences of land use changes for metal mobility and risk assessment. Soil Science, v.163, p.859-871, 1998.
- RUSSEL, J.D.; PARFITT, R.L.; FRASER, A.R.; FARMER, V.C. Surface structures of gibbsite, goethite and phosphated goethite. Nature, v.248, p.220-221, 1974.
- SALET, R.L. Toxidez de alumínio no sistema de plantio direto. Porto Alegre: UFRGS/FA, 1998. 109p. (Tese - Doutorado).
- SCHINDLER, P.W.; GAMSJAGER, H. Acid-base reactions of the TiO2 (anatase)-water interface and the point of zero charge of TiO2 suspensions. Kolloid-Zeitschrift und Zeitschrift fur Polymere, v.250, p.759-763, 1972.
- SCHINDLER, P.W.; FUERST, B.; DICK, R.; WOLF, P.U. Ligand properties of surface silanol groups. I. Surface complex formation with Fe3+, Cu2+, Cd2+ and Pb2+ Journal of Colloid and Interface Science, v.55, p.469-475, 1976.
- SIGG, L.; STUMM, W. The interaction of anions and weak acids with the hydrous goethite (a-FeOOH) surface. Colloids Surface, v.2, p.101-117, 1981.
- SILVA, É.T.; MELO, W.J.; TEIXEIRA, S.T. Chemical attributes of a degraded soil after application of water treatment sludges. Scientia Agricola, v.62, p.559-563, 2005.
- SILVA, F.R.; FERREYRA H.F.F.; COELHO, M.A.; AQUINO, B.F. Efeito da relação de adsorção de sódio sobre o coeficiente de seletividade Na-Ca em solos aluviais de diferentes texturas. Revista Brasileira de Ciência do Solo, v.15, p.9-13, 1991.
- SILVEIRA, M.L.A.; ALLEONI, L.R.F. Copper adsorption in tropical Oxisols. Brazilian Archives of Biology and Technology, v.46, p.529-536, 2003.
- SINGH, U.; UEHARA, G. Electrochemistry of the double layer: principles and applications to soils. In: SPARKS, D.L. (Ed.). Soil physical chemistry Boca Raton: CRC Press, 1986. p.2-38.
- SOARES, M.R. Coeficiente de distribuição (Kd) de metais pesados em solos do estado de São Paulo. Piracicaba: USP/ESALQ, 2004. 213p. (Tese - Doutorado).
- SPATHARIOTIS, E.; KALLIANOU, C. Adsorption of copper, zinc, and cadmium on clay fraction of two acid soils: Surface complexation modeling. Communications in Soil Science and Plant Analysis, v.32, p.3185-3205, 2001.
- SPOSITO, G. Derivation of the freundlich equation for ion-exchange reactions in soils. Soil Science Society of America Journal, v.44, p.652-654, 1980.
- SPOSITO, G. The chemistry of soils New York: Oxford University Press, 1989. 277p.
- STUMM, W. Chemistry of the solid-water interface New York: John Wiley & Sons, 1992. 428p.
- STUMM, W.; HUANG, C.P.; JENKINS, S.R. Specific chemical interaction affecting the stability of dispersed systems. Croatian Chemical Acta, v.42, p.223-245, 1970.
- STUMM, W.; HOHL, J.; DALANG, F. Interaction of metal ions with hydrous oxide surfaces. Croatian Chemical Acta, v.48, p.491-504, 1976.
- STUMM, W.; KUMMERT, R.; SIGG, L. A ligand exchange model for the adsorption of inorganic and organic ligands at hydrous oxide surfaces. Croatian Chemical Acta, v.53, p.291-312, 1980.
- TESSENS, E.; SHAMSHUDDIN, J. Characteristics related to charges in Oxisols of Peninsular Malaysia. Pedologie, v.32, p.85-106, 1982.
- UEHARA. G; GILLMAN, G. The mineralogy, chemistry, and physics of tropical soils with variable charge Boulder: Westview Press. 1981. 170p. (Westview Tropical Agricultural Series, 4).
- USEPA (Environmental Protection Agency). Soil screening guidance: thecnical background document. Washington DC: EPA, Office of Solid Waste and Emergency Response, 1996. 168p.
- WANN, S.S.; UEHARA, G. Surface charge manipulation in constant surface potential soil colloids: I. Relation to sorbed phosphorus. Soil Science Society of America Journal, v.42, p.565-570, 1978.
- WHITE, R.E. Principles and practice of Soil Science The soil as a natural resource. 3.ed. Oxford: Blackwell Science, 1997. 348p.
- WILD, A. Nitrate leaching under bare fallow at a site in Northern Nigeria. Journal of Soil Science, v.23, p.315-324, 1972.
- WONG, M.T.F.; HUGHES, R.; ROWELL, D.L. The retention of nitrate in acid soils from the tropics. Soil Use and Management, v.6, p.72-74, 1990.
- YATES, D.E.; LEVINE, S.; HEALY, T.W. Site-binding model of the electrical double layer at the oxide/water interface. Journal of the Chemical Society Faraday Transactions, v.70, p.1807-1818, 1974.
Publication Dates
-
Publication in this collection
22 Dec 2006 -
Date of issue
Dec 2006
History
-
Accepted
15 Nov 2006 -
Received
01 Aug 2006

