Abstract
Transition state theory (TST) is the most widely used formalism for theoretical calculations of diffusion coeffcients of defects in solids. In this work, we test its validity for the case of vacancy diffusion in silicon. The diffusion coefficient directly obtained from molecular-dynamics simulations with a classical (Stillinger-Weber) potential is compared with TST predictions. Our results confirm the validity of TST for this system.
Vacancy Diffusion in Silicon: Analysis of Transition State Theory
R. R. Gattass, Belita Koiller, and R. B. Capaz
Instituto de Física, Universidade Federal do Rio de Janeiro,
Caixa Postal 68528, Rio de Janeiro, RJ 21945-970, Brazil
Received February, 1998
Transition state theory (TST) is the most widely used formalism for theoretical calculations of diffusion coeffcients of defects in solids. In this work, we test its validity for the case of vacancy diffusion in silicon. The diffusion coefficient directly obtained from molecular-dynamics simulations with a classical (Stillinger-Weber) potential is compared with TST predictions. Our results confirm the validity of TST for this system.
Diffusion coefficient calculations for defects in solids are almost exclusively done in the framework of transition state theory (TST) [1]. Within this formalism, the defect jump rate G is written as
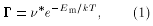
where the migration energy Em is determined by the energy difference between two absolutely static atomic configurations: The ground state and the saddle-point, i.e.,

The saddle-point is the configuration of maximum energy along the adiabatic reaction pathway. The pre-factor n* can be seen as an effective "jump-attempt" frequency, and it is given within TST by
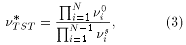
where the products in the numerator and denominator are over all normal mode frequencies of the ground state and saddle-point, respectively.
In this work, we test the validity of TST predictions for the specific case of vacancy diffusion in silicon. Vacancy in silicon has been and still is [2] one of the most studied point defects in semiconductor physics, partly due to its role in mediating impurity diffusion. Our calculations are based on a classical Stillinger-Weber [3] (SW) interatomic potential. This potential is chosen mainly for its simplicity but, as shown below, it provides a reasonable agreement with more sophisticated methods for many calculated properties.
The system is modeled by a supercell with periodic boundary conditions and we start out by searching for its ground state. A vacancy is initially formed by removing one of the silicon atoms in the supercell, without relaxing the remaining atoms. This unrelaxed configuration is found to be a metastable state for the SW potential. The true ground state is associated with a 0.56 Å inward relaxation of the vacancy first neighbors, and it is shown in Fig. 1(a) (or Fig. 1(c)). Our result using a classical potential is in reasonable agreement with recent ab initio calculations [4] which indicate an inward relaxation of 0.4 Å.
Fragment of the supercell around the vacancy, illustrating the diffusion pathway: (a) initial ground state, (b) saddle-point (split-vacancy) and (c) final ground state.
The saddle-point configuration can be guessed from symmetry arguments and is known as split-vacancy. The resulting migration pathway is then indicated in Fig. 1, which shows the initial, saddle and final configurations. In this pathway, one of the vacancy neighbors moves to an intermediate position midway its initial location and the vacancy position.
The formation energies for the vacancy and split-vacancy calculated for a 64-site cell (thus containing 63 silicon atoms) are shown in Table 1 , along with the corresponding ab initio and experimental results. The TST migration energy, calculated as in Eq. (2) are also given. Notice that TST migration energy (0.49 eV) is in reasonable agreement with the experimental values of 0.18-0.45 eV [7]. Convergence with respect to the supercell size was investigated for 216-, 512- and 1000-site cells, with the migration energy converging to 0.54 eV.
Formation energies for the ground state (E0) and saddle-point (Es) and migration energy (Em) for the vacancy in silicon (in eV).
We now proceed to the calculation of the pre-factor n*. The fully relaxed configurations for a 64-site cell are used as input for the calculation of the vibration spectra at both the ground state and saddle-point. The matrix of force constants is obtained by a finite-difference approach, in which the matrix element Fia,jb is approximated by 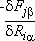 , where dFjb is the calculated force on atom j, direction b, for a small displacement dRia on atom i along direction a starting from equilibrium. This matrix is then diagonalized and the pre-factor n* is calculated as prescribed in Eq. (3). We find n* = 1.1 ×1013 Hz.
, where dFjb is the calculated force on atom j, direction b, for a small displacement dRia on atom i along direction a starting from equilibrium. This matrix is then diagonalized and the pre-factor n* is calculated as prescribed in Eq. (3). We find n* = 1.1 ×1013 Hz.
The accuracy of TST predictions is tested by comparison with results directly obtained from molecular dynamics (MD) simulations. We perform MD simulations within the constant NEV ensemble, where N is the number of Si atoms, V is the volume and E is the total energy. The atomic trajectories are generated from a 5th-order predictor-corrector algorithm with a time step of 10-16 s. Due to the small time steps required for accurate dynamics, the amount of elapsed time is limited. In the simulations presented here, we typically simulate diffusion for 10-9 s.
The diffusion coefficient is obtained from the simulations through:

where
i is the position of atom i at time t. Since the mean square displacement can be related to the number of jumps n in the diamond lattice by

where a is the lattice parameter, the jump rate is given by:

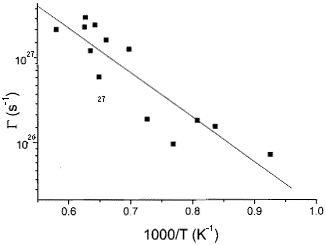
Results for G at different temperatures are shown in Fig. 2 as an Arrhenius plot. From the linear fit of the data according to Eq. (1), we obtain n* = 2 ±1 ×1013 Hz and Em = 0.44±0.07 eV for the frequency and migration energy, respectively. These results are to be compared with the TST predictions: n*TST = 1.1 ×1013 Hz and 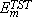 = 0.49 eV.
= 0.49 eV.
Jump rate G as a function of the inverse temperature from molecular dynamics simulations for the 64-site system. The filled line represents the best fit from which the dynamical values for the n* and Em are extracted.
These results show no clear discrepancy between TST predictions TST and MD simulations, specially for the migration energy. For the pre-factor n*, the TST value barely fits the large error bars. Indeed, pre-factors are expected to be more affected by two of the key approximations in TST: the harmonic approximation and the neglect of multiple jumps and return jumps. The harmonic approximation is used in TST in order to perform analytically the integrations needed to determine the ratio of partition functions that will lead to Equations (1-3) [1]. This approximation can be, in some cases, improved through numerical integration [8]. The second approximation is a more dramatic problem for TST. Multiple jumps and return jumps occurring in a time scale much smaller than the mean residence time are correlated events not easily introduced in the framework of TST. Even though there have been some attempts in this direction [9], this remains an open, difficult and important problem in defect physics. We plan to address these issues in a future work.
Acknowledgments
Work partially supported by Conselho Nacional de Desenvolvimento Cientí fico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) and Programa de Apoio a Núcleos de Excelência do Ministério de Ciência e Tecnologia (PRONEX-MCT).
- [1] G. H. Vineyard, J. Phys. Chem. Solids 3, 121 (1957).
- [2] A. Antonelli, E. Kaxiras, and D. J. Chadi, Phys. Rev. Lett. 81, 2088 (1998).
- [3] F. H. Stillinger and T. A. Weber, Phys. Rev. B 31, 5262 (1985).
- [4] H. Seong and L. J. Lewis, Phys. Rev. B 53, 9791 (1996).
- [5] P. J. Kelly and R. Car, Phys. Rev. B 45, 6543 (1992).
- [6] S. Dannefaer, P. Mascher, and D. Kerr, Phys. Rev. Lett. 56, 2195 (1986).
- [7] G. D. Watkins, in Deep Centers in Semiconductors, edited by S. T. Pantelides (Gordon and Breach, New York, 1986).
- [8] M. J. L. Sangster and A. M. Stoneham, J. Phys. C 17, 6093 (1984).
- [9] C. P. Flynn and G. Jacucci, Phys. Rev. B 25, 6225 (1982).
Publication Dates
-
Publication in this collection
23 Feb 2001 -
Date of issue
Dec 1999
History
-
Received
Feb 1998