
Abstract
In this work we present a theoretical study on electron scattering by both polar and nonpolar polyatomic molecules in the low-energy range. More specifically, we report calculated elastic and rotationally inelastic differential cross sections for electron scattering by CH4, H2O, and H2S in the (2.14-30)-eV range. Exact static-exchange plus model correlation-polarization potentials are used to represent the electron-molecule interaction. The Schwinger variational iterative method is used to solve the scattering equations. In addition, the adiabatic-nuclei-rotation approximation is applied to calculate rotational cross sections. The comparison of our calculated results with experimental and other theoretical data available in the literature is encouraging.
a23v32n3
Elastic and Rotational Excitation Cross Sections for Electron Scattering by Polyatomic Molecules
L. E. Machado*, L. M. Brescansinf, and M.-T. Lee
* Departamento de Física, UFSCar, 13565-905, São Carlos, SP, Brazil
Instituto de Física "Gleb Wataghin'', UNICAMP, 13083-970, Campinas, SP, Brazil
Departamento de Química, UFSCar, 13565-905, São Carlos, SP, Brazil
Received on 10 December, 2001. Revised version received on 7 March, 2002.
In this work we present a theoretical study on electron scattering by both polar and nonpolar polyatomic molecules in the low-energy range. More specifically, we report calculated elastic and rotationally inelastic differential cross sections for electron scattering by CH4, H2O, and H2S in the (2.14-30)-eV range. Exact static-exchange plus model correlation-polarization potentials are used to represent the electron-molecule interaction. The Schwinger variational iterative method is used to solve the scattering equations. In addition, the adiabatic-nuclei-rotation approximation is applied to calculate rotational cross sections. The comparison of our calculated results with experimental and other theoretical data available in the literature is encouraging.
I Introduction
Electron scattering by molecules is a very wide field of research. In particular, collisions of electrons with small polyatomic molecules such as CH4, H2O and H2S among others, play an important role in several areas of investigation [1], mainly in atmospheric and plasma physics, radiation biology, chemistry, etc. In the low-energy range, both rotationally elastic and rotationally inelastic processes are known to be relevant for studies in some of these areas [2,3].
Besides this intrinsic interest in rotational processes, another interesting point arises for the case of electron collisions with polar targets. In view of the smallness of the electron mass, as compared to that of the nuclei, and due to the fact that the collision time is typically much shorter than the characteristic times of the nuclear motions, theoretical studies often rely on the fixed-nuclei framework, which consists in treating the nuclei as fixed-in-space particles during the collision process. However, this treatment of electron scattering by polar molecules is known to lead to divergent differential cross sections (DCS) in the forward direction, due to the slow falloff of the T-matrix elements for large angular-momentum partial-wave components [4]. This divergence can be removed only by the introduction of the nuclear motion in the Hamiltonian [5]. That inclusion, in principle, will require the solution of complicated coupled rovibronic equations [2]. But, indeed, that divergence can be avoided in a much simpler manner, by calculating rotationally-summed cross sections obtained by adding up rotationally-resolved elastic and inelastic cross sections, calculated in the framework of the adiabatic-nuclei-rotation (ANR) approximation [6]. Also, due to the dipolar nature of such molecules, it is very interesting to study both the influence of the long-range dipole interaction on the behavior of the angular distributions for elastic and inelastic processes and the efficiency of the inelastic collisions with electrons that can excite the rotor target states.
Although absolute cross sections for rotationally unresolved elastic scattering of electrons by CH4, H2O and H2S are known since the last decades of the past century, the lack of high-resolution apparatuses has made rotational excitation measurements much more scarce. To our knowledge, experimental results for rotationally resolved DCS are available only for H2O [3] and CH4 [7]. On the theoretical side, rotational excitation cross sections for some low-lying rotational excited levels have been reported for CH4 [6,8-13], H2O [6,13-16] and H2S [13,17,18]. However, significant discrepancies have been observed in the reported cross sections for the (0®1) rotational excitation for H2O [6,13] and H2S [13,17,18] as well as for the (0®3) and (0®4) rotational excitations for CH4 [6,9,11]. Therefore, additional theoretical studies on rotational excitation processes of these targets using a different formulation would be useful.
In this paper we report calculated DCS for rotationally elastic and inelastic scattering of electrons by H2O, H2S and CH4 for incident energies ranging from 2.14 to 30 eV. Also, rotationally-summed DCS at 10 and 20 eV are reported and compared with some existing unresolved experimental and theoretical results [19-31]. A static-exchange-polarization (SEP) approximation is used to calculate accurate low angular-momentum partial-wave scattering amplitudes via the Schwinger variational iterative method (SVIM) [32]. Rotationally elastic and inelastic cross sections are calculated within the framework of the ANR approximation. For polar targets, higher angular-momentum dipole-potential T-matrix components were then added to infinity through a first Born approximation (FBA) closure formula. This procedure was previously used by our and other groups [6,11,33,34] for studies of elastic electron-polar molecule scattering.
The organization of the paper is as follows. In Sec. II we present a very brief discussion of the theory we have used and report some details of the calculations. In Sec. III we compare our results to experimental and other theoretical data available in the literature. Finally, in Sec. IV we summarize our conclusions.
II Theory and calculation
In this section we will briefly discuss the method used. A more detailed discussion on the SVIM can be found elsewhere [32]. Within the ANR framework, the DCS for the excitation of an asymmetric-top rotor (as H2O and H2S) from an initial rotational level J 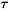 to a final level J¢ ¢ is given by
to a final level J¢ ¢ is given by

where fJ
M ® J¢¢M¢ is the rotational excitation scattering amplitude, related to the rotational eigenfunctions of the target by

In Eq. (1) kJ and kJ¢¢ are the magnitudes of the linear momenta of the incident and the scattered electron, respectively. In Eq. (2) W º (a, b, g) are the Euler angles defining the orientation of the molecular principal axes [35] and f LF is the electronic part of the laboratory-frame (LF) scattering amplitude which can be related to the corresponding body-frame (BF) T matrix by an usual frame transformation. The eigenfunctions YJ

where the symmetric-top eigenfunctions are given by

where
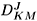
are the well-known Wigner rotation matrices [35]. For a spherical-top rotor (as CH
4) the rotational eigenfunctions are still given by Eq. (4), but the degeneracy in both
K and
M quantum numbers reduces Eq. (1) to the form:

The Lippmann-Schwinger scattering equation for elastic electron-molecule collision is solved using the SVIM, in which the continuum wavefunctions are single-centre expanded as

where the superscripts (+) and (-) denote the incoming-wave and outgoing-wave boundary conditions, respectively. The BF T matrix can also be conveniently partial-wave expanded as

where
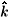
0 and
are the linear-momentum directions of the incident and scattered electrons in the BF, respectively. In Eqs. (6) and (7)
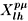
(
) are the symmetry-adapted functions [37] which are expanded in terms of the usual spherical harmonics as follows:

Here
p is an irreducible representation (IR) of the molecular point group, m is a component of this representation and
h distinguishes between different bases of the same IR corresponding to the same value of
l. The coefficients
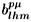
satisfy important orthogonality relations and are tabulated for
C
2v and
Oh point groups [37].
In actual calculations, the expansions in Eqs. (6) and (7) have been truncated in a set of cutoff parameters. For polar targets (H2O and H2S), higher partial-wave contributions were taken into account via a Born-closure procedure which is the same as used by Rescigno and Lengsfield [33] and by our group [34] for the study of the elastic scattering of electrons by water. A BF Born-closure formula for the T matrix can be written as:

where  are the partial-wave T-matrix elements calculated via SVIM and truncated to some cutoff values L = (lc, hc),
are the partial-wave T-matrix elements calculated via SVIM and truncated to some cutoff values L = (lc, hc),  are the corresponding partial-wave point-dipole FBA T-matrix elements and T B is the complete point-dipole FBA T matrix.
are the corresponding partial-wave point-dipole FBA T-matrix elements and T B is the complete point-dipole FBA T matrix.
In this work the electron-molecule scattering dynamics is represented by an optical potential, given by

where Vst, Vex, and Vcp are the static, the exchange, and the correlation-polarization contributions, respectively.
In our calculation, Vst and Vex are derived exactly from a Hartree-Fock SCF target wavefunction. A parameter-free model potential introduced by Padial and Norcross [38] is used to account for the correlation-polarization contribution. In this model, a short-range correlation potential between the scattering and the target electrons is defined in an inner interaction region and a long-range polarization potential in an outer region. The correlation potential is calculated by a free-electron-gas model, derived using the target electronic density according to Eq. (9) of Padial and Norcross [38]. In addition, the asymptotic form of the polarization potential is used for the long-range electron-target interaction. The first crossing of the correlation and polarization potential curves defines the inner and the outer regions. No further adjustable parameters are needed for the calculation of Vcp.
The SCF wavefunctions of the targets are calculated from standard contracted basis sets [39]. At the experimental equilibrium geometries, the calculated SCF total energies are -40.1987, -76.0199, and -398.68 a.u., for CH4, H2O, and H2S, respectively, to be compared with the corresponding values of -40.2155 [26], -76.0632 [40], and -398.61 a.u. [41]. The polarizabilities a0 = 17.5 a.u. (for CH4) [42], a0 = 24.55 a.u. (for H2S) [42], and a0 = 10.6253 a.u.,  = -0.6363 a.u. and
= -0.6363 a.u. and  = 0.30788 a.u. (for H2O) [43] were used to calculate the asymptotic form of Vcp. All partial-wave expansions were truncated, at most, at lc = 16 with all possible values of h £ l retained for a given l. For the assumed lc, it has been verified that all dipole and the corresponding point-dipole FBA T-matrix elements agree within 5%. Our results shown below were all converged within four iterations.
= 0.30788 a.u. (for H2O) [43] were used to calculate the asymptotic form of Vcp. All partial-wave expansions were truncated, at most, at lc = 16 with all possible values of h £ l retained for a given l. For the assumed lc, it has been verified that all dipole and the corresponding point-dipole FBA T-matrix elements agree within 5%. Our results shown below were all converged within four iterations.
III Results and discussion
Although the main interest of the present work is to discuss rotational excitation cross sections of polyatomic molecules, some calculations of elastic (rotationally summed) cross sections were also performed. These cross sections were obtained by adding the rotationally resolved DCS , given by Eq. (1), up to convergence. For illustration, representative results of DCS for electron scattering by CH4, H2O, and H2S are shown in Figs.1-3, respectively, for incident energies of 10 and 20 eV. Some selected experimental [19-23, 28-30] and theoretical results [24-27, 31] are also presented for comparison. Our calculated DCS agree well with the experimental results and also with most calculated data. Rotationally resolved cross sections for each of these molecules are discussed separately below.

III.1 e--CH4 scattering
Figures 4-6 show our results for rotationally resolved DCS at 5, 7.5 and 10 eV, respectively, along with the experimental results of Müller et al. [7] and the theoretical results of Jain and Thompson [6], Abusalbi et al. [8], Brescansin et al. [9], and Varella et al. [13], when available. Comparing our data with other theoretical results, a general qualitative agreement with each other is observed. In contrast, the recent calculated DCS of Gianturco et al. [11] (not shown) have presented unphysical oscillations for the (0®3) and (0® 4) transitions. Quantitatively, our rotationally resolved DCS are in very good agreement with those of Abusalbi et al. [8] at 10 eV and they are also in fair agreement with the other calculated results for this energy. Also, a good agreement is seen among the present and experimental results for the (0® 0) rotational transition, as well as for the (0®3) transition at 5 eV and the (0® 4) transition at 10 eV. For the other cases, the agreement is still fair. Nevertheless, none of the calculations was able to reproduce the sharp minimum at around 125º for the (0®3) transition and the strong decrease at large scattering angles (q ³ 120º) for the (0®4) transition in the experimental values of the DCS at 7.5 eV [7]. Since the data reported by Müller et al. [7] are the only experimental results available in the literature, new measurements would help clarify these discrepancies.

III.2 e--H2O scattering
Figures 7(a) and 7(b) show our results for the rotationally elastic and the averaged  [(0®1)+(1®0)] rotational excitation/de-excitation DCS at 2.14 eV, respectively, along with the experimental results of Jung et al. [3] and the available theoretical results of Jain and Thompson [6] and Gianturco et al. [16]. Similar comparison, but for 6 eV, is shown in Figs. 7(c) and 7(d). Our results agree reasonably well with the experimental and other theoretical results, although none of the calculations was able to predict the broad maximum centered at around 70º seen in the experimental data of Fig. 7(d). Again, since the results of three different calculations agree reasonably well with each other at least qualitatively, we think that new measurements would be needed to confirm or to disprove that structure.
[(0®1)+(1®0)] rotational excitation/de-excitation DCS at 2.14 eV, respectively, along with the experimental results of Jung et al. [3] and the available theoretical results of Jain and Thompson [6] and Gianturco et al. [16]. Similar comparison, but for 6 eV, is shown in Figs. 7(c) and 7(d). Our results agree reasonably well with the experimental and other theoretical results, although none of the calculations was able to predict the broad maximum centered at around 70º seen in the experimental data of Fig. 7(d). Again, since the results of three different calculations agree reasonably well with each other at least qualitatively, we think that new measurements would be needed to confirm or to disprove that structure.
Figures 8(a-d) show our DCS for the (0®0,1,2,3) rotational transitions, respectively, at 30 eV. Unfortunately, there are no corresponding experimental results available in the literature. Therefore, comparison is made only with the other calculated data [13,16]. A general overall agreement is observed among all the calculated results.
III.3 e -H2S scattering
In Figs. 9 and 10 we present the (0®0,1,2,3) rotational transition cross sections, for incident energies of 10 and 15 eV, respectively. The calculated results of Varella et al. [13] and Gianturco [18] (for 0®1 only) are also shown for comparison. General good agreement is found, both in shape and magnitude, between all calculations, except for the (0®1) excitation, where a significant disagreement between our data and the SMC results can be seen. As this discrepancy is only apparent in the excitation channel for which the Born-closure procedure is needed (J¢ = 1), it could be due to different implementations of this procedure in the calculations. Unfortunately, no experimental result is available in order to shed some light on this discussion. Anyway, our results are in better agreement with the calculated data of Gianturco [18].
IV Summary and conclusions
In this work, we report a theoretical study of low-energy-electron scattering by small polyatomic molecules. Rotationally elastic and inelastic, as well as rotationally summed cross sections are presented in comparison with some existing experimental and other theoretical results. Our calculated rotationally summed DCS are in general good agreement with the measured rotationally unresolved data. For some cases, our rotationally resolved results also agree quite well with the corresponding experimental data. This good agreement supports the description of the interaction dynamic considered in the present study and the methods used for solving the scattering equations. Extension to higher incident energies and other molecular targets are underway.
References
[1] S. Trajmar, D. F. Register, and A. Chutjian, Phys. Rep. 97, 219 (1983).
[2] N. F. Lane, Rev. Mod. Phys. 59, 29 (1980).
[3] K. Jung, Th. Antoni, R. Müller, K-H. Kochem, and H. Erhardt, J. Phys. B 15, 3535 (1982).
[4] D. W. Norcross and L. A. Collins, Advances in Atomic and Molecular Physics 18, 341 (1982).
[5] L. A. Collins and D. W. Norcross, Phys. Rev. A 18, 467 (1978).
[6] A. Jain and D. G. Thompson, J. Phys. B 16, 3077 (1983).
[7] R. Müller, K. Jung, K.-H. Kochem, W. Sohn, and H. Erhardt, J. Phys. B 18, 3971 (1985).
[8] N. Abusalbi, R. A. Eades, T. Nam, D. Thirumalai, D. A. Dixon, D. G. Truhlar, and M. Dupuis, J. Chem. Phys. 78, 1213 (1983).
[9] L. M. Brescansin, M. A. P. Lima, and V. McKoy, Phys. Rev. A 40, 5577 (1989).
[10] A. Jain, Z. Phys. D 21, 153 (1991).
[11] F. A. Gianturco, J. A. Rodriguez-Ruiz, and N. Sanna, Phys. Rev. A 52, 1257 (1995).
[12] M. T. do N. Varella, M. H. F. Bettega, and M. A. P. Lima, Z. Phys. D 39, 59 (1997).
[13] M. T. do N. Varella, M. H. F. Bettega, A. P. P. Natalense, L. G. Ferreira, and M. A. P. Lima, Braz. J. Phys. 31, 21 (2001).
[14] F. A. Gianturco, J. Phys. B 24, 3837 (1991).
[15] R. Greer and D. G. Thompson, J. Phys. B 27, 3533 (1994).
[16] F. A. Gianturco, S. Meloni, P. Paioletti, R. R. Lucchese, and N. Sanna, J. Chem. Phys. 108, 4002 (1998).
[17] A. Jain and D. G. Thompson, J. Phys. B 17, 443 (1983).
[18] F. A. Gianturco, J. Phys. B 24, 4627 (1991).
[19] H. Tanaka, T. Okada, L. Boesten, T. Suzuki, T. Yamamoto, and M. Kubo, J. Phys. B 15, 3305 (1982).
[20] L. Vuskovic and S. Trajmar, J. Chem. Phys. 78, 4947 (1983).
[21] P. J. Curry, S. Newell, and A. C. Smith, J. Phys. B 18, 2303 (1985).
[22] T. W. Shyn and T. E. Cravens, J. Phys. B 23, 293 (1990).
[23] L. Boesten and H. Tanaka, J. Phys. B 24, 821 (1991).
[24] A. Jain, Phys. Rev. A 34, 3707 (1986).
[25] P. McNaughten, D. G. Thompson, and A. Jain, J. Phys. B 23, 2405 (1990).
[26] T. Nishimura and Y. Itikawa, J. Phys. B 27, 2309 (1994).
[27] M. H. F. Bettega, A. P. P. Natalense, M. A. P. Lima, and L. G. Ferreira, J. Chem. Phys. 103, 10566 (1995).
[28] T. W. Shyn and S. Y. Cho, Phys. Rev. A 36, 5138 (1987).
[29] W. M. Johnstone and W. R. Newell, J. Phys. B 24, 3633 (1991).
[30] R. J. Gulley, M. J. Brunger, and S. J. Buckman, J. Phys. B 26, 2913 (1993).
[31] B. H. Lengsfield III, T. N. Rescigno, C. W. McCurdy, and S. Parker (1992), as quoted by Ref. 30.
[32] R. R. Lucchese, G. Raseev and V. McKoy, Phys. Rev. A 25, 2572 (1982).
[33] T. N. Rescigno and B. H. Lengsfield III, Z. Phys. D 24, 117 (1992).
[34] L. E. Machado, M.-T. Lee, L. M. Brescansin, M. A. P. Lima, and V. McKoy, J. Phys. B 28, 467 (1995).
[35] A. R. Edmonds, Angular Momentum and Quantum Mechanics, Princeton University Press, Princeton (NJ), (1960).
[36] C. Van Winter, Physica XX, 274 (1954); A. Jain and D. G. Thompson, Comput. Phys. Commun. 30, 301 (1983).
[37] P. G. Burke, N. Chandra, and F. A. Gianturco, J. Phys. B 5, 2212 (1972).
[38] N. T. Padial and D. W. Norcross, Phys. Rev. A 29, 1742 (1984).
[39] T. H. Dunning Jr., J. Chem. Phys. 55, 716 (1971).
[40] T. H. Dunning Jr., R. M. Pitzer, and S. Aung, J. Chem. Phys. 57, 5044 (1972).
[41] I. Cacelli, V. Caravetta, A. Rizzo, and R. Moccia, J. Chem. Phys. 102, 1230 (1995).
[42] J. O. Hirschfelder, C. F. Curtis, and R. B. Bird, Molecular Theory of Gases and Liquids, John Willey, New York (NY), (1954).
[43] N. S. Hush and M. L. Williams, Chem. Phys. Lett. 6, 163 (1970).
- [1] S. Trajmar, D. F. Register, and A. Chutjian, Phys. Rep. 97, 219 (1983).
- [2] N. F. Lane, Rev. Mod. Phys. 59, 29 (1980).
- [3] K. Jung, Th. Antoni, R. Müller, K-H. Kochem, and H. Erhardt, J. Phys. B 15, 3535 (1982).
- [4] D. W. Norcross and L. A. Collins, Advances in Atomic and Molecular Physics 18, 341 (1982).
- [5] L. A. Collins and D. W. Norcross, Phys. Rev. A 18, 467 (1978).
- [6] A. Jain and D. G. Thompson, J. Phys. B 16, 3077 (1983).
- [7] R. Müller, K. Jung, K.-H. Kochem, W. Sohn, and H. Erhardt, J. Phys. B 18, 3971 (1985).
- [8] N. Abusalbi, R. A. Eades, T. Nam, D. Thirumalai, D. A. Dixon, D. G. Truhlar, and M. Dupuis, J. Chem. Phys. 78, 1213 (1983).
- [9] L. M. Brescansin, M. A. P. Lima, and V. McKoy, Phys. Rev. A 40, 5577 (1989).
- [10] A. Jain, Z. Phys. D 21, 153 (1991).
- [11] F. A. Gianturco, J. A. Rodriguez-Ruiz, and N. Sanna, Phys. Rev. A 52, 1257 (1995).
- [12] M. T. do N. Varella, M. H. F. Bettega, and M. A. P. Lima, Z. Phys. D 39, 59 (1997).
- [13] M. T. do N. Varella, M. H. F. Bettega, A. P. P. Natalense, L. G. Ferreira, and M. A. P. Lima, Braz. J. Phys. 31, 21 (2001).
- [14] F. A. Gianturco, J. Phys. B 24, 3837 (1991).
- [15] R. Greer and D. G. Thompson, J. Phys. B 27, 3533 (1994).
- [16] F. A. Gianturco, S. Meloni, P. Paioletti, R. R. Lucchese, and N. Sanna, J. Chem. Phys. 108, 4002 (1998).
- [17] A. Jain and D. G. Thompson, J. Phys. B 17, 443 (1983).
- [18] F. A. Gianturco, J. Phys. B 24, 4627 (1991).
- [19] H. Tanaka, T. Okada, L. Boesten, T. Suzuki, T. Yamamoto, and M. Kubo, J. Phys. B 15, 3305 (1982).
- [20] L. Vuskovic and S. Trajmar, J. Chem. Phys. 78, 4947 (1983).
- [21] P. J. Curry, S. Newell, and A. C. Smith, J. Phys. B 18, 2303 (1985).
- [22] T. W. Shyn and T. E. Cravens, J. Phys. B 23, 293 (1990).
- [23] L. Boesten and H. Tanaka, J. Phys. B 24, 821 (1991).
- [24] A. Jain, Phys. Rev. A 34, 3707 (1986).
- [25] P. McNaughten, D. G. Thompson, and A. Jain, J. Phys. B 23, 2405 (1990).
- [26] T. Nishimura and Y. Itikawa, J. Phys. B 27, 2309 (1994).
- [27] M. H. F. Bettega, A. P. P. Natalense, M. A. P. Lima, and L. G. Ferreira, J. Chem. Phys. 103, 10566 (1995).
- [28] T. W. Shyn and S. Y. Cho, Phys. Rev. A 36, 5138 (1987).
- [29] W. M. Johnstone and W. R. Newell, J. Phys. B 24, 3633 (1991).
- [30] R. J. Gulley, M. J. Brunger, and S. J. Buckman, J. Phys. B 26, 2913 (1993).
- [31] B. H. Lengsfield III, T. N. Rescigno, C. W. McCurdy, and S. Parker (1992), as quoted by Ref. 30.
- [32] R. R. Lucchese, G. Raseev and V. McKoy, Phys. Rev. A 25, 2572 (1982).
- [33] T. N. Rescigno and B. H. Lengsfield III, Z. Phys. D 24, 117 (1992).
- [34] L. E. Machado, M.-T. Lee, L. M. Brescansin, M. A. P. Lima, and V. McKoy, J. Phys. B 28, 467 (1995).
- [35] A. R. Edmonds, Angular Momentum and Quantum Mechanics, Princeton University Press, Princeton (NJ), (1960).
- [36] C. Van Winter, Physica XX, 274 (1954);
- A. Jain and D. G. Thompson, Comput. Phys. Commun. 30, 301 (1983).
- [37] P. G. Burke, N. Chandra, and F. A. Gianturco, J. Phys. B 5, 2212 (1972).
- [38] N. T. Padial and D. W. Norcross, Phys. Rev. A 29, 1742 (1984).
- [39] T. H. Dunning Jr., J. Chem. Phys. 55, 716 (1971).
- [40] T. H. Dunning Jr., R. M. Pitzer, and S. Aung, J. Chem. Phys. 57, 5044 (1972).
- [41] I. Cacelli, V. Caravetta, A. Rizzo, and R. Moccia, J. Chem. Phys. 102, 1230 (1995).
- [42] J. O. Hirschfelder, C. F. Curtis, and R. B. Bird, Molecular Theory of Gases and Liquids, John Willey, New York (NY), (1954).
- [43] N. S. Hush and M. L. Williams, Chem. Phys. Lett. 6, 163 (1970).
Publication Dates
-
Publication in this collection
04 Dec 2002 -
Date of issue
Sept 2002
History
-
Received
10 Dec 2001 -
Reviewed
07 Mar 2002









