Resumos
As borrachas, para serem convertidas em materiais úteis do ponto de vista de aplicações, precisam ser misturadas a alguns ingredientes, os quais vão lhes dar as características desejadas para, então, serem vulcanizadas. Neste artigo, os aspectos históricos da vulcanização são abordados bem como a evolução dos aditivos essenciais à maioria das formulações.
Vulcanização; borracha natural; aceleradores; enxofre
Rubbers, to be turned into useful materials from the commercial applications point of view, need the addition of a few other ingredients which, upon vulcanization, will impart the desired characteristics to the material. In this paper, historical aspects of vulcanization are revised as well as the evolution of the additives used in most of the formulations.
Vulcanization; natural rubber; accelerators; sulfur
ARTIGO DE DIVULGAÇÃO
Aspectos históricos da vulcanização
Historical aspects of vulcanization
Helson M. da CostaI; Leila L. Y. VisconteI; Regina C. R. NunesI; Cristina R. G. FurtadoII
IIMA, UFRJ
IIGrupo de Polímeros, Instituto de Química, UERJ
RESUMO
As borrachas, para serem convertidas em materiais úteis do ponto de vista de aplicações, precisam ser misturadas a alguns ingredientes, os quais vão lhes dar as características desejadas para, então, serem vulcanizadas. Neste artigo, os aspectos históricos da vulcanização são abordados bem como a evolução dos aditivos essenciais à maioria das formulações.
Palavras-chave: Vulcanização, borracha natural, aceleradores, enxofre.
ABSTRACT
Rubbers, to be turned into useful materials from the commercial applications point of view, need the addition of a few other ingredients which, upon vulcanization, will impart the desired characteristics to the material. In this paper, historical aspects of vulcanization are revised as well as the evolution of the additives used in most of the formulations.
Keywords: Vulcanization, natural rubber, accelerators, sulfur.
Aspectos históricos
A borracha faz parte de um grupo de materiais industriais conhecidos como materiais de engenharia, que inclui também metais, fibras, concreto, madeira, plásticos, vidros, dos quais depende parte da tecnologia moderna[1].
Os índios americanos foram os primeiros a descobrir e fazer uso das propriedades singulares da borracha. Os aventureiros espanhóis, que sucederam Colombo no princípio do século XVI, os encontraram praticando um jogo organizado com uma bola que saltava melhor do que qualquer coisa conhecida na Europa, até então. Durante os cem anos que se seguiram, os europeus descobriram, gradativamente, uma série de outras utilizações que os índios davam a este extraordinário material. Eles o espalhavam em roupas para torná-las impermeáveis, moldavam-no em forma de argila para produzir uma espécie primitiva de botina, ou em vasilhames flexíveis e seringas, e também ofereciam-no a seus deuses, como incenso[1,2].
Entretanto, este material apresentava dois grandes problemas: os usuários encontravam dificuldades em trabalhar com a borracha sólida e os artefatos tornavam-se moles e pegajosos quando submetidos ao calor. Em tempo frio, tornavam-se progressivamente duros e rígidos, até que no rigor do inverno, tornavam-se quase completamente inflexíveis. Além disso, desenvolviam odores desagradáveis após um período curto de tempo[1-3].
Cerca de 450 anos depois, em torno de 1800, esse material ganhou aceitação universal em função da descoberta do processo de vulcanização. Vulcanização, palavra derivada da mitologia romana (Vulcano, Deus do fogo e do trabalho com metais), é o termo usado para descrever o processo através do qual a borracha reage com enxofre para produzir uma rede de ligações cruzadas entre as cadeias poliméricas. Se um número suficiente de ligações cruzadas é formado, o artefato adquire uma forma fixa, não mais moldável, porém ainda flexível e elástica. Se, no entanto, muitas ligações cruzadas são formadas, o elastômero é convertido em um sólido rígido[1-3].
A descoberta da vulcanização é atribuída a Charles Goodyear, nos Estados Unidos, e a Thomas Hancock, na Inglaterra. Ambos desenvolveram patentes em 1840. A vulcanização da borracha provocava uma melhora pronunciada nas propriedades químicas e físicas, em relação ao material não vulcanizado. Não havia mais o amolecimento do material em temperaturas elevadas ou o congelamento em contato com o frio, além de torná-lo mais resistente quimicamente. Embora os artefatos de borracha desenvolvidos a partir das formulações de Goodyear e Hancock fossem superiores em muitos aspectos, quando comparados com a borracha não vulcanizada, eles ainda estavam longe do ideal. Grandes quantidades de enxofre e tempos de cura relativamente longos eram necessários[1-3]. Além disso, a reversão, processo pelo qual se produz uma marcante deterioração das propriedades físicas, era um problema sério na época. Os vulcanizados apresentavam coloração intensa, indesejada migração do enxofre para a superfície (afloramento) e exibiam muito pouca resistência ao envelhecimento.
Hoje é sabido que a rede de ligações cruzadas formada pela vulcanização sem aceleradores ou em presença de aceleradores inorgânicos (em geral óxidos metálicos tais como de zinco, cálcio, magnésio ou chumbo) é muito complexa. Além dos diferentes tipos de ligações cruzadas com enxofre, os vulcanizados contêm uma grande proporção de modificações na cadeia principal tais como: ciclizações sulfídicas, insaturações conjugadas, e isomerização cis/trans da dupla ligação. A vulcanização com enxofre em ausência de aceleradores é, portanto, um processo ineficaz[1-3].
O passo mais importante com relação à química da vulcanização ocorreu com a descoberta dos aceleradores orgânicos, em 1900. Além de aumentarem a velocidade de vulcanização, esses aditivos trouxeram muitas outras vantagens. O uso de aceleradores permitiu o emprego de temperaturas mais baixas e tempos de cura menores. Conseqüentemente, não houve mais a necessidade de submeter a borracha a condições drásticas e, desse modo, a possibilidade de degradação térmica e oxidativa foi minimizada. Além disso, o nível de enxofre pode ser reduzido e, ainda assim, sem prejuízo para as propriedades físicas do vulcanizado. O resultado foi a redução do afloramento de enxofre e a maior resistência ao envelhecimento. A possibilidade de reversão também foi reduzida. Vulcanizados transparentes ou coloridos puderam ser preparados. Negro de fumo e outras cargas foram incorporados na mistura para melhorar as propriedades físicas do produto final, sem afetar drasticamente a velocidade da reação de cura. Por fim, a rede de ligações cruzadas derivada da vulcanização em presença de aceleradores orgânicos mostrou ser mais simples e com menos modificações do que a rede produzida somente com o enxofre, daí o processo ser chamado de eficiente[3-5].
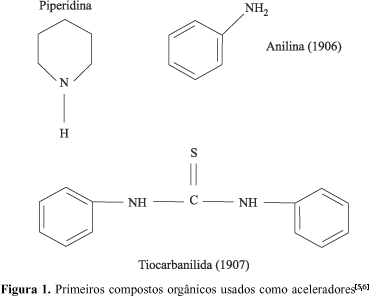
A primeira patente para aceleradores orgânicos foi desenvolvida pela firma alemã Bayer e cobria o uso de aminas alifáticas, cicloalifáticas e hetero cíclicas. Em comum com muitas importantes descobertas, os químicos William Wolfgang e Walter Oswald, testando bases orgânicas para possível ação antioxidante, observaram o efeito acelerador da piperidina. Eles perceberam que a adição de bases orgânicas fortes aumentava em muito a velocidade de vulcanização. Independentemente dos químicos alemães, Karl Oenslager, da Diamond Rubber Co., já havia descoberto aceleradores orgânicos em 1906, mas publicou suas pesquisas somente depois da Primeira Guerra Mundial. Oenslager tinha inicialmente usado anilina mas, devido à toxicidade, substituiu-a pela tiocarbanilida e por outros compostos menos tóxicos (Figura 1)[5,6].
Houve um grande avanço no campo dos aceleradores químicos após a Primeira Guerra Mundial. Três classes principais de aceleradores foram desenvolvidas e ainda hoje são largamente usadas. Estas classes são: (1) aceleradores baseados em guanidinas; (2) aceleradores baseados em sais metálicos e de amônio do ácido ditiocarbâmico; e (3) aceleradores baseados no 2-mercaptobenzotiazol[5,6].
Somente três tipos de aceleradores do tipo guanidina estão disponíveis comercialmente (Figura 2), dos quais o mais importante é a N,N'-difenilguanidina (DPG). Embora por algum tempo estes tipos de aceleradores tenham sido muito populares, eles são agora usados principalmente como aceleradores secundários, em conjunto com aceleradores primários tais como 2-mercaptobenzotiazol (MBT) e seus derivados[5,6].
Os ditiocarbamatos (Figura 3), os quais são classificados como ultra-aceleradores, tornaram-se proeminentes em torno de 1918. De fato, a vulcanização com enxofre acelerada por dialquilditiocarbamato de amônio (ADADC) é tão rápida que ocasiona problemas sérios de "scorch" (vulcanização prematura durante a composição). Tentativas de moderar a extraordinária rapidez do ADADC resultaram na descoberta dos sais metálicos, especialmente o sal de zinco (ZDADC), o qual promovia a cura em velocidade muito mais lenta. Contudo, a vulcanização com ZDADC é ainda muito rápida e não resolve totalmente os problemas de controle do "scorch". Já os sulfetos de tiuram como, por exemplo, dissulfeto de tetrametiltiuram (TMTD), monossulfeto de tetrametiltiuram (TMTM), e tetrassulfeto de dipentametilenotiuram (DPMTT), foram resultado de posterior pesquisa para substituir o enxofre do grupo mercaptan do ácido ditiocarbâmico a fim de obter-se uma velocidade de cura mais satisfatória. Sulfetos de tiuram tornaram-se uma classe muito importante de aceleradores (Figura 3). No mesmo período (1920) foi descoberto que o óxido de zinco aumentava a ação dos aceleradores orgânicos e, desde então, este tem sido considerado como imprescindível em composições elastoméricas, exercendo o papel de ativador. Junto com o óxido de zinco, ácido esteárico tem sido usado em muitras composições como co-ativador, embora seu mecanismo de atuação ainda não tenha sido elucidado. O fato é que o uso de estearato de zinco no lugar do óxido de zinco e do ácido esteárico não produz o mesmo efeito ativador[5-7].
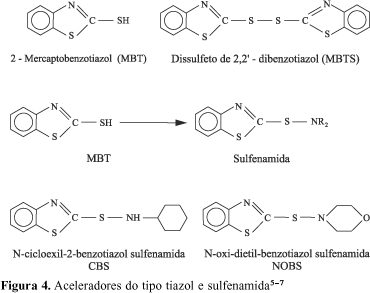
Aceleradores baseados no 2-mercaptobenzotiazol (MBT) e seus derivados (Figura 4) são, sem dúvida, a mais importante classe de aceleradores usados industrialmente. O desenvolvimento dos aceleradores baseados no MBT foi atribuído simultaneamente a C. W. Bedford e L. B. Sebrell, nos Estados Unidos, e a G. Bruni e E. Romani na Itália, em 1920. Estes químicos descobriram que o MBT, o derivado dissulfeto (MBTS), e os sais metálicos (sal de zinco, ZMBT), eram aceleradores muito efetivos e com importantes vantagens sobre os derivados do ácido ditiocarbâmico. A maior vantagem que os aceleradores do tipo MBT oferecem diz respeito ao maior tempo para a pré-cura, o que é um fator de excepcional importância na manufatura de artefatos de borracha[5-7].
Esforços para otimizar o aumento do tempo de pré-cura dos aceleradores do tipo MBT levaram ao desenvolvimento, em 1932, de um importante subgrupo de aceleradores as sulfenamidas (Figura 4). O enxofre do grupo mercaptan presente no MBT foi substituído por uma amina e essa substituição resultou em um retardo da vulcanização. Esses materiais provaram ser muito valiosos e são, nos dias de hoje, provavelmente, os aceleradores mais largamente usados. Um resumo dos avanços na química dos aceleradores pode ser encontrado na Figura 5[6,7].
O avanço seguinte na química da vulcanização somente ocorreu em 1968 com a introdução da N-cicloexiltioftalimida (CTP, Santogard PVI), com a finalidade de promover a inibição da vulcanização prematura. Esse produto proporcionou, pela primeira vez, o controle do tempo de pré-cura, sem interferir na velocidade de cura o problema de "scorch" estava resolvido. Com os avanços tecnológicos que ocorriam desde 1937, a disponibilidade de antidegradantes do tipo amina e novas variedades de negro de fumo, menos onerosas e igualmente reforçantes, a formulação para a manufatura de uma borracha estava completa[5-8].
Para elastômeros insaturados, um sistema típico de vulcanização com enxofre é composto pelo enxofre, um óxido metálico (geralmente óxido de zinco), um ácido graxo (geralmente ácido esteárico), e um ou mais promotores orgânicos. A reação ocorre em uma faixa de temperatura de 150 ± 30 °C. Para elastômeros saturados, a vulcanização é conduzida através de radicais livres gerados por peróxidos, compostos halogenados ou radiação de alta energia[5-8].
Os fatores a serem considerados na seleção dos sistemas de vulcanização devem incluir o tipo de elastômero, o tipo e a quantidade de óxido de zinco e ácido graxo (a eficiência do ácido esteárico como co-ativador na vulcanização deve-se à sua solubilidade no elastômero, peso molecular e ponto de fusão), a velocidade de vulcanização, a resistência requerida à fadiga e às condições de serviço. Freqüentemente, misturas de aceleradores são usadas. Tipicamente, um acelerador do tipo benzotiazol é usado juntamente com pequenas quantidades de um do tipo ditiocarbamato (tiuram) ou amina. O efeito da combinação de dois tipos diferentes de aceleradores é sinérgico, ou seja, um acelerador ativa o outro, e melhores velocidades de formação de ligações cruzadas são obtidas. Misturas de aceleradores do mesmo tipo geram resultados intermediários[5-8].
Aumento nas concentrações de enxofre e acelerador gera altas densidades de ligações cruzadas e, portanto, alto módulo de elasticidade, rigidez, dureza, etc. Contudo, com o aumento da razão entre as concentrações acelerador/enxofre, a proporção de ligações monossulfídicas aumenta em composições de borracha. Grandes quantidades de acelerador (com relação ao enxofre) também levam a abundantes grupos pendentes do tipo Sx Ac, o qual está ligado nas cadeias de borracha. Por outro lado, altas razões entre as concentrações de enxofre/acelerador geram mais ligações polissulfídicas e mais enxofre combinado com a borracha, na forma de anéis heterocíclicos de seis membros, distribuídos ao longo das cadeias. Além disso, ligações olefínicas duplas conjugadas ao longo da cadeia polimérica também aparecem[7-9].
A rota geral do processo de vulcanização é descrita pelo Esquema 1. Primeiro, um complexo ativo do acelerador é formado através de uma interação preliminar entre o acelerador e o ativador, na presença de zinco solúvel. Esse complexo pode reagir com o enxofre molecular, mediante a abertura do anel de S8, para formar um agente sulfurante[10]. A existência desses complexos foi detectada quimicamente[11,12] e também pela combinação das análises de NMR/HPLC[13].
Posteriormente, o agente sulfurante pode reagir com as cadeias de borracha para formar um precursor de ligações cruzadas. O precursor foi detectado por evidências experimentais[14,15] na forma de um polissulfeto, ligado a um fragmento da molécula do acelerador, e está presente como grupo pendente na cadeia principal da borracha. O precursor, subseqüentemente, leva à formação de ligações cruzadas polissulfídicas.
Neste meio tempo, a diminuição da eficiência na formação de ligações cruzadas pode ocorrer devido a reações laterais como decomposição ou dessulfurização dos precursores[14,16]. Por causa destas reações laterais, a formação de sulfetos cíclicos, dienos conjugados, trienos, ZnS, e grupos monossulfídicos pendentes pode ser observada. Estas espécies não são capazes de contribuir para a geração de ligações cruzadas. Foi observado que a atividade, a concentração do complexo zinco-acelerador[15,17] e a temperatura são as principais variáveis de controle para as possíveis reações laterais citadas.
Finalmente, a rede de ligações cruzadas inicialmente formada sofre maturação e, durante esse processo, a dessulfurização (diminuição do tamanho das ligações cruzadas, eventualmente levando a ligações monossulfídicas) e/ou a decomposição das ligações cruzadas polissulfídicas ocorrem[18,19].

Conclusão
Embora a evolução dos aceleradores e de outros aditivos fundamentais para o processo de vulcanização, seja com enxofre ou com outro agente de cura (peróxidos, óxidos metálicos e outros), esteja bem documentada na literatura, a vulcanização é um tema que está longe de ter sido esgotado.
O assunto suscita interesse sobretudo pela investigação contínua das possíveis reações pelas quais os diferentes aditivos da composição (ativadores, aceleradores e agentes de cura) interagem e como tais interações afetam a rede de ligações cruzadas. Neste contexto, vários métodos são citados para a caracterização estrutural das composições vulcanizadas com enxofre e para a elucidação dos mecanismos de reação envolvidos, tais como:
• Uso de sondas químicas ("chemical probes"), as quais reagem seletivamente com elementos estruturais específicos sendo possível determinar a concentração das ligações cruzadas (mono-, di- e polissulfídicas), bem como a quantidade total de enxofre combinado e a quantidade de modificações na cadeia principal do elastômero. Este método é baseado na seletividade e reatividade dos reagentes usados e constitui uma das técnicas mais antigas para análise dos vulcanizados, como citado por Saville e Watson[20].
• Vulcanização de compostos modelo, os quais possuem boa correlação com sistemas elastoméricos reais, além de serem relativamente fáceis de ser analisados. Exemplos de compostos modelo são o 2-metil-2-penteno, o 2,3-dimetil-2-buteno, o 1-deceno, o 2,3-dimetil-1-buteno, entre outros.
• Técnicas espectroscópicas como a ressonância magnética nuclear de 13C no estado sólido, a qual em combinação com outras técnicas, tem permitido superar as dificuldades de análise de estruturas vulcanizadas por métodos convencionais, como a espectrometria no infravermelho ou no ultravioleta.
• Métodos termoanalíticos tais como: a análise térmica diferencial (DTA) e a calorimetria diferencial de varredura (DSC). Tais técnicas podem ser úteis na investigação do mecanismo de reação, da solubilidade dos ingredientes em uma borracha em particular e da reatividade de uma dada borracha. A desvantagem reside em como fazer uma interpretação detalhada diante da complexidade química da vulcanização, além da identificação de quais fenômenos são de natureza química e/ou física. Esta abordagem foi aplicada para o estudo da vulcanização do poliisopreno em uma série de artigos de Kruger et al.[21-25].
A presença de cargas em composições elastoméricas também é um fator adicional a ser considerado. Cargas como a sílica influenciam de modo adverso a vulcanização e exigem que novos ingredientes sejam incorporados às formulações, tais como aminas ou glicóis. Por conseguinte, todo o mecanismo das reações que levam à vulcanização é afetado. Neste contexto, estudos envolvendo a influência dessas cargas comerciais assim como de cargas alternativas, as quais tem se mostrado substitutos potenciais para a sílica[26-29], sobre a cinética de vulcanização de composições de borracha natural estão sendo conduzidos e os resultados serão reportados oportunamente.
Recebido: 06/09/02
Aprovado: 12/02/03
Autor para correspondência: Regina C. R. Nunes, IMA, UFRJ, Caixa Postal 68525, CEP: 21945-970, Rio de Janeiro, RJ. E-mail: rcnunes@ima.ufrj.br
-
11. Borracha Sintética – A história de uma indústria, International Institute of Synthetic Rubber Producers Inc., Holanda (1973).
- 2. Stern, H. J. "History", in: Rubber Technology and Manufacture, cap.1, C. M. Blow (ed), Newnes-Butterworths, London (1975).
- 3. Shreve, R. N. & Brink Jr., J. A. "Indústrias da borracha", in: Indústria de Processos Químicos, cap.36, Horácio Macedo, Editora Guanabara (1977).
- 4. Subramaniam, A. "Natural rubber", in: Rubber Technology, cap.4, M. Morton (ed.), Van Nostrand Reinhold Company Inc., New York (1987).
- 5. Coleman, M. M.; Shelton, J. R. & Koening, J. L. Ind. Eng. Chem., Prod. Res. Develop., 13 (3), p.154 (1974).
- 6. Coran, A. Y. Chemtech, february, p.106 (1983).
- 7. Morell, S. H. "The chemistry and technology of vulcanization", in: Rubber Technology and Manufacture, cap.5, C. M. Blow, Newnes-Butterworths, London (1975).
- 8. Brabin, W. W. & Rodgers, M. B. "The Science of rubber compounding", in: The Science and Technology of Rubber, cap.9, F. R. Eirich & B. Erman, Academic Press, New York (1994).
- 9. Coran, A. Y. "Vulcanization", in: The Science and Technology of Rubber, cap.7, F. R. Eirich & B. Erman, Academic Press, New York (1994).
- 10. Bateman, L.; Moore, C. G.; Porter, B. & Saville, B. "Vulcanization", in: The Chemistry and Physics of Rubber-Like Substances, cap.15, Bateman L. (ed.), Maclaren and Sons Ltd., London (1963).
- 11. Campbell, R. H. & Wise, R. W. Rubber Chem. Technol., 37, p.635 (1964).
- 12. Coran, A. Y. Rubber Chem. Technol., 37, p.679 (1964).
- 13. Krejsa, M. R.; Koening, J. L. & Sullivan, A. B. Rubber Chem. Technol., 67, p.348 (1994).
- 14. Campbell, D. S. J. Appl. Polym. Sci., 14, p.1409 (1970).
- 15. Parks, C. R.; Parker, D. K.; Chapman, D. A. & Cox, W. L. Rubber Chem. Technol., 43, p.572 (1970).
- 16. Morrison, N. J. & Porter, M. Rubber Chem. Technol., 57, p.63 (1984).
- 17. Parks, C. R.; Parker, D. K. & Chapman, D. A. Rubber Chem. Technol., 45, p.467 (1972).
- 18. Milligan, B. Rubber Chem. Technol., 39, p.1115 (1966).
- 19. Loo, C. T. Polymer, 15, p.357 (1974).
- 20. Saville, B.; Watson, A. A. - Rubber Chem. Technol., 40, p.100 (1967).
- 21. Kruger, F. W. H.; McGill, W. J. - J. Appl. Polym. Sci., 42, p.2643 (1991).
- 22. Kruger, F. W. H.; McGill, W. J. - J. Appl. Polym. Sci., 42, p.2661 (1991).
- 23. Kruger, F. W. H.; McGill, W. J. - J. Appl. Polym. Sci., 44, p.581 (1992).
- 24. Kruger, F. W. H.; McGill, W. J. - J. Appl. Polym. Sci., 45, p.573 (1992).
- 25. Kruger, F. W. H.; McGill, W. J. - J. Appl. Polym. Sci., 45, p.749 (1992).
- 26. Gomes, R. C. M.; Visconte, L. L. Y.; Russi, C. G. F.; Nunes, R. C. R. ; Furtado, C. J. A. Revista de Química Industrial, 68, p. 15 (2000).
- 27. Da Costa, H. M.; Nunes, R. C. R.; Visconte, L. L. Y.; Furtado, C. R. G. Kautschuk Gummi Kunststoffe, 54, p. 242 (2001)
- 28. Sereda, L.; Pereira, H. L.; Nunes, R. C. R.; Visconte, L. L. Y.; Furtado, C. R. G. - Kautschuk Gummi Kunststoffe, 54, p. 474 (2001)
- 29. Da Costa, H. M.; Visconte, L. L. Y.; Nunes, R. C. R.; Furtado, C. R. G. J. Applied Polym. Sci., 83, p. 2331 (2002)
Datas de Publicação
-
Publicação nesta coleção
01 Ago 2003 -
Data do Fascículo
Jun 2003
Histórico
-
Aceito
12 Fev 2003 -
Recebido
06 Set 2002