Abstract
The ionic conductivity, thermal stability and lithium transference number have been investigated in composite polymer electrolytes (CPE) of poly(vinylidene fluoride - hexafluoropropylene) (PVDF-HFP) incorporating nano fillers of AlO[OH]n or gammaAl2O3, with LiClO4 as salt. The compatibility of the composite polymer electrolyte with lithium metal anode has also been studied. The effect of particle size on the compatibility of Li/ CPE/ Li symmetric cells has also been analyzed.
Ionic conductivity; compatibility; composite polymer electrolyte; transference number; thermal stability
ARTIGO TÉCNICO CIENTÍFICO
Electrochemical studies on inert filler incorporated poly (vinylidene fluoride hexafluoropropylene) (PVDF HFP) composite electrolytes
J. WilsonI; G. RaviI; M. Anbu KulandainathanII
IAlagappa University, India
IICentral Electrochemical Research Institute, India
Corresponding author Corresponding author: M. Anbu Kulandainathan Central Electrochemical Research Institute Karaikudi 630 006, India Email: manbu123@yahoo.com
ABSTRACT
The ionic conductivity, thermal stability and lithium transference number have been investigated in composite polymer electrolytes (CPE) of poly(vinylidene fluoride - hexafluoropropylene) (PVDF-HFP) incorporating nano fillers of AlO[OH]n or gAl2O3, with LiClO4 as salt. The compatibility of the composite polymer electrolyte with lithium metal anode has also been studied. The effect of particle size on the compatibility of Li/ CPE/ Li symmetric cells has also been analyzed.
Keywords: Ionic conductivity, compatibility, composite polymer electrolyte, transference number, thermal stability.
Introduction
Large research efforts have been devoted on the development of lithium batteries with polymer electrolytes as it is expected to offer high energy density, flexible and safe batteries for domestic and defence applications[1-2]. Although polymer electrolytes based on poly ethylene oxide [PEO] was invented by P.V. Wright in 1973, their technological importance was realized only in early 1980's. Nevertheless, PEO- based electrolytes is one of the earliest and most extensively studied and, they offer conductivity of the order ranging from 10-8 to 10-4 S cm-1 at temperature between 40 and 100 ºC, that excludes for practical application[3]. The ionic conductivity[4], transference number[5,6], DSC and NMR[7], thermal[8], polarization[9] and cycling performance[10,11] of PEO and copolymer of P(EOMMA)-based electrolytes[12] have been reported. Poly acrylonitrile (PAN) based electrolytes offer high ionic conductivity (10-3 S cm-1) at room temperature, good electrochemical stability (< 4.5V) and an appreciable transference number (0.6). However, these electrolytes undergo severe passivation upon contact with lithium metal anode[13-14].
Ijiima et al.[15] and Scrosati et.al.[16] have explored the possibility of using PMMA as polymer host. The ionic conductivity of PMMA remains very close to that of liquid electrolyte. Studies have also been made on PVC-based electrolytes[17,18]. Recently, Stephan et.al.[19,20] have attempted to enhance the mechanical strength of PMMA electrolytes by blending them with PVC.
Very recently, PVdF as a host has drawn the attention of many researchers due to its appealing properties. It has high anodic stability due to strong electron withdrawing functional group and has a high dielectric constant (e = 8.4) which helps in a greater ionization of lithium salts[21]. Unfortunately, PVdF-based polymer electrolytes suffer due to syneresis; a phenomenon by which the liquid component separates out from the host matrix in due course or upon application of pressure which leads to battery leak and related safety problems. They also have poor mechanical properties. These films have to be hardened either by chemical or physical curing, which leads to high processing costs. Very recently, by virtue of its appealing properties PVdF-HFP has drawn the attention of many researchers. The copolymer of vinylidene fluoride with hexafluoro propylene P(VdF-HFP) which contain amorphous domains capable of trapping large amount of liquid electrolytes and the crystalline phase acts as a mechanical support[22].
Recent studies reveal that only composite polymer electrolytes incorporated with inorganic filler can offer mechanically strong and safe batteries with improved electrolyte- electrode compatibility[23-27]. One of the functions of the fillers is the reduction of crystallinity of the host matrix. Most of the recent papers deals with PEO incorporated with inert fillers [26,27]. In the present study, an attempt has been made to study the effect of inorganic filler, AlO[OH]n or g- Al2O3 on the ionic conductivity and compatibility of PVdF-HFP copolymer electrolytes. In order to evaluate the possible applications of the various classes of polymer electrolytes, there is a definite need for a better understanding of these interfacial characteristics. With this aim, we have undertaken a systematic investigation of the behavior of passivation phenomena displayed by the Li electrode in contact with various composite polymer electrolytes.
Experimental
Poly (vinlylidene fluoride hexafluoro propylene)(PVdF-HFP) (Kynar, Japan) and lithium perchlorate (LiClO4) (Aldrich, USA) were dried under vacuum at 90 ºC for 12h before use. The inert fillers, AlO[OH]n and g- Al2O3 of particle size 14nm and 5 microns respectively were also dried at 120 ºC for 12h before use. The preparation of nano composite electrolyte involved the dispersion of the selected inert filler and LiClO4 salt in anhydrous tetrahydrofuran (THF), followed by the addition of PVdF-HFP as depicted in table 1 and the resultant solution was cast as film. The solvent was allowed to evaporate and the composite film was obtained and the average thickness of the film was 30-50 µm. This procedure yielded homogenous and mechanically strong membranes, which were dried under vacuum at 45-60 ºC for 24 h.
The prepared films were sandwiched between the two stainless steel discs of diameter 1 cm and the ionic conductivity of the membranes was measured using an electrochemical impedance analyzer (IM6 - Bio Analytical Systems, USA) in the 50 mHz to 100 kHz frequency range at various temperatures viz., 0, 10, 20, 30, 45, 60 and 75 ºC. The lithium ion transference number, T+Li was estimated using the method proposed by Vincent and co-workers[29]. Watanabe et.al.[30], have also proposed a method for the determination of lithium ion transference number. The values of T+Li were measured by imposing a dc polarization pulse to a cell of the lithium- composite polymer electrolyte lithium type and by following the time evolution of the resulting current flow using the expression[7]

This method consists of measuring the ac impedance and by dc chronoamperometry respectively. The resistance and the current cross a symmetrical Li/CPE/Li cells polarized by a dc voltage pulse, V. In the present study the dc voltage pulse applied to the cell was 10mV. The measurements were taken the initial time of the applied dc voltage pulse (t=to, R=Ro, l=lo) and under steady conditions (t=ts, R=Rs, l=ls).
The compatibility of the Li/CPE/Li symmetric cells was investigated by studying the time dependence of the impedance of the systems. In the present study sample, S5 was used as it exhibited maximum ionic conductivity.
Results and Discussion
Ionic conductivity
XRD analysis
Figure 1(a) and (b) display the XRD pattern of PVdF-HFP polymer and PVdF-HFP- AlO [OH]n- LiClO4 sample. The peaks at 2q = 18.2, 20, 26.6 and 38, which, correspond to (1 0 0) (0 2 0), (1 1 0) and (0 2 1) crystalline peaks of PVdF. This confirms the partial crystallization of PVdF units in the copolymer and gives a semi- crystalline structure of PVdF-HFP[33]. A similar pattern was observed when g-Al2O3 was incorporated in the polymer host. The crystallinity of the polymer has been considerably decreased upon the addition of the inert filler and lithium salt. It is quite obvious from the Figure 1(b) that, the intensity of the crystalline peaks decreases and broadens. This reduction in crystallinity upon the addition of inert filler is attributed to small particles of inert fillers which changes the chain re-organization and facilitates for higher ionic conduction[34].
Ionic conductivity
Figure 2 and 3 depict the ionic conductivity as a function of filler concentration, [AlO(OH)]n in the polymer host for fixed lithium salt content (5%), LiClO4 with g-Al2O3 and AlO[OH] respectively. It is clear from the figure that the ionic conductivity has considerably been increased (up to three order) in both cases upon the addition of inert filler. The ionic conductivity of the inert filler system is higher than the undoped system for all the temperatures studied. The ionic conductivity increases with the increase of filler content up to 15% and decreases further even when the concentration of the filler was increased. These results are in accordance with those reported earlier in which Al2O3 was used as filler in PEO- based electrolytes[35]. As commonly found in composite materials, the conductivity is not linear function of the filler concentration. At low concentration levels the diffusion effect which, tends to depress the conductivity, is effectively contrasted by the specific interactions of the ceramic surfaces, which promotes fast ion transport and follows the Arrhenius conductivity. Hence an apparent enhancement in conductivity is seen in both cases. At higher filler content, the dilution effect predominates and the conductivity is lowered. On the other hand, when the concentration of the filler was increased the dilution effect predominates and the conductivity decreases[36]. Thus the maximum conductivity is achieved only in the concentration region of 8-15 wt%. According to Scrosati and co-workers[37] from their NMR studies that the local dynamics of the lithium ions, in particular lithium mobility, is not changed by filler which supports the idea that the enhancement of conductivity by adding a filler is caused by stabilizing and increasing the fraction of amorphous phase. Our XRD result also substantiates this point. However, indeed, this point does not hold good solely for the enhancement of conductivity where the polymer has amorphous phase by its own nature. According to Scrosati et al.[35], the Lewis acid groups of the added inert filler (eg. OH group of the added both fillers ) may compete with the Lewis- acid lithium cations for the formation of complexes with the PEO chains as well as the anions of the added lithium salt. Subsequently, this results a structural modifications on the filler surfaces, due to the specific actions of the polar surface groups of the inorganic filler. The Lewis acid-base interaction centers with the electrolytic species, thus lowering the ionic coupling and promotes the salt dissociation via a sort of "ion-filler complex" formation. Indeed, this effect could be the reason for the observed enhanced conductivity for both systems studied[35].
A similar trend was observed for the films comprising AlO[OH]n as filler (Figure 3). While comparing the ionic conductivity of the films containing g - Al2O3 and AlO[OH]n, the films which possess AlO[OH]n as filler exhibits higher ionic conductivity than the films with g - Al2O3. This increased ionic conductivity of AlO[OH]n films are attributed to the smaller particle size of the filler. According to Scrosati et al.[32] the nano sized inert fillers incorporated polymers exhibits higher ionic conductivity due to the enhanced amorphous phase nature of the polymer hosts.
Compatibility studies
Lithium is an attractive anode material for secondary batteries that provides a larger capacity of 3800 mAhg-1. This capacity is about ten times higher than that of a carbon based anode (376 mAhg-1) with a composition of LiC6[38]. However, due to the extreme reactivity of the lithium metal, most of the developed polymer electrolytes passivate lithium. Passivation of lithium electrode in a non- aqueous electrolyte is a well studied phenomenon; however lithium- solid polymer electrolyte interfacial study is progressing now. Indeed there are preliminary, but convincing results that lithium passivation phenomena, similar to those commonly experienced in liquid electrolytes, also occur in polymer electrolyte cells. However, the nature and the mechanism of the passivation phenomena are still unclear and under debate and one can only postulate that they depend in a non- predictable way on the type of electrolyte involved, the temperature of operation and the residual impurities[39,40].
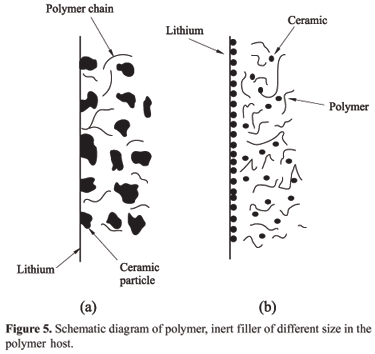
Figure 4 shows the evolution of interfacial resistance "Ri" as a function of time of the symmetric cell, Li/ CPE/Li, comprising PVdF-HFP-LiClO4- g-Al2O3 and (PVdF-HFP)- LiClO4 AlO[OH]n. It is quite obvious from the figure, that the composite polymer electrolyte with AlO[OH] as filler offers minimum interfacial resistance. The value of "Ri" increases and decreases in an irregular way with time and after 180 hours the value remains more or less stable. This may be attributed by assuming the morphology of the passivation films changes with time to a finally acquire a non-compact possibly porous structure. As depicted in Figure 5, the inert particles depending upon the volume fraction would tend to minimize the area of lithium electrode exposed to polymers containing O, OH- species and thus reduce the passivation process. It is also foreseeable that smaller size particles would impart an improved performance as compared to larger size particles because they will cover more surface area[23]. The formation of an insulated layer of ceramic particles at the electrode surface is probable at higher volume fraction of a passive ceramic phase. This insulating layer will impede electrode reactions. This may very well have happened when excessive amount of the passive ceramic phase were introduced into the polymer matrix.
Transference number
The transport number of an ion in a given electrolyte solution is defined as the fraction of the total electrical current carried in the solution by that particular ion. Every species will have a transport number ti and must be always positive and the value lies between zero and one.

However in practice it is impossible to distinguish between simple ions and other charged species. Therefore the observable quantity depends on the transfer of an ion constituent and not a free ion. This transference number is based on transport of the gram equivalent one faraday is called transference number.

Several methods have been employed to measure the transference number of solid polymer electrolytes. The most commonly used method was introduced Vincent et al.[20]
In order to substantiate the conductivity results further, we have measured the Li+ transference number, tLi+, for all the samples and are displayed in Table 2. This table reports the results in terms of numerical values of tLi+. The transference number values may equally be affected by the interfacial properties with lithium metal anode also[41]. An apparent increase in the transference number, tLi+ (Table 1) is observed when passing from the filler- free to the filler incorporated composite electrolytes. This further supports the ionic conductivity results.
TG- DTA analysis
In order to find the thermal stability of the composite polymer electrolytes the samples were subjected to TG/DTA analysis. Figure 6 shows the TG-DTA traces of PVdF-HFP, PVdF-HFP/ g-Al2O3 / LiClO4 and PVdF-HFP/ AlO[OH]n / LiClO4 respectively. It is evident from Figure 6(a), there is an endothermic peak observed at around 130 ºC, which indicates the melting point of HFP[42]. Around 425 ºC an irreversible decomposition process takes place, which indicates the decomposition of PVdF matrix[33]. This indicates that PVdF-HFP is stable up to 120 ºC. On the other hand, the film, which possesses g-Al2O3 and LiClO4, a weight loss takes place around 130 ºC also corresponds to an exothermic peak, indicates the decomposition of lithium salt and HFP components. Further another weight loss was observed (about 5%) at around 325 ºC with a corresponding exothermic peak indicates the decomposition of complex film. As mentioned before the decomposition of PVdF matrix takes place at around 425 ºC.
More interestingly, film with AlO[OH]n, shows a weight loss of about 7% around 135 ºC and further another weight loss of about 3% around 370 ºC which indicates the decomposition of AlO[OH]n. Further the decomposition of PVdF-matrix takes place at around 425 ºC.
Conclusions
The following conclusions were drawn on the basis of the above studies: PVdF-HFP composite polymer electrolytes were prepared with two different inert fillers, namely: AlO[OH]n and g- Al2O3 with lithium salt LiClO4. The composite polymer electrolyte with AlO[OH]n offers better compatibility. The nano particle sized inert fillers offer better interfacial resistance than with micron- sized particles. These films are thermally stable up to 120ºC, which is the desired temperature for the operation of composite polymer batteries for electric vehicle applications.
Enviado: 18/04/05
Reenviado: 13/07/05
Aprovado: 19/08/05
- 1. Fray, F. M. - Solid polymer electrolytes - Fundamentals and Technological Applications, VCH (1993)
- 2. Scrosati, B.- Applications of Electroactive polymers, London: Chapman and Hall (1993).
- 3. Bertheir, C.; Gorecki, W.; Minier, M.; Armand, M. B.; Chanbagno, J. M. & Rigaud, P. - Solid State Ionics 11, p.91 (1983).
- 4. Gray, F. M.; MacCallum & Vincent, C. A. - Solid State Ionics, 18-19, p.225 (1986).
- 5. Bouridah, A.; Dalard, F.; Daroo, D.; Armand, M. B. - Solid State Ionics, 18-19, p.287 (1986).
- 6. Cameroon, G. G.; Harvie, J. A. & Ingram, M. D. - Solid State Ionics 34, p.65 (1986).
- 7. Gorecki, W.; Andreani, R.; Bertheir, C.; Armand, M.; Mali, M.; Roos, J. & Brinmann D - Solid State Ionics 18-19, p.295 (1986).
- 8. Chiodelli, G.; Ferlon, P.; Magistrio, A. & Saner, M. - Solid State Ionics 28-30, p.1009 (1988).
- 9. Bruce, P. G. & Vincent, C. A. - Solid State Ionics 40-41 p.607 (1990).
- 10. Bonino F.; Ottavini, M.; Scrosati, B. & Pistoia, G. - J. Electrochem. Soc., 135, p.12 (1988).
- 11. Croce, F. & Scrosati, B. - J. Power Sources 43-44, p.9 (1993).
- 12. Kim, D. W. - J. Power Sources 87, p.78, 87:78 (2000).
- 13. Abraham, K. M. & Alamgir, M. - J. Electrochem. Soc., 137, p.1657 (1990)
- 14. Manuel Stephan, A.; Thirunakaran, R.; Renganathan, N. G.; Muniyandi, N, & Ramesh Babu, B. - In: Radhakrishna Editor, Trends in Materials Science, Vol 1. Allied Publishers, p. 99
- 15. Iijima, Y.; Tyoguchi, Y. & Eda, N. - Denki Kagaku, 53, p.619 (1985).
- 16. Appetecchi, G. B,; Croce, F. & Scrosati, B. - Elecrochim. Acta, 40, p.991 (1995).
- 17. Abraham, K. M. & Alamgir, A. - J. Electrochem. Soc., 140, p.L96 (1993).
- 18. Mary Sukeshini, A.; Nishimoto, A. & Watanabe. M. - Solid State Ionics, 86-88, p.385 (1996).
- 19. Manuel Stephan, A.; Thirunakaran, R,; Renganathan, N. G.; Pitchumani, S.; Muniyandi, N. & Rama moorthy S, - J. Power Sources, 81- 82, p.752 (1999).
- 20. Manuel Stephan, A.; Prem Kumar, T.; Thirunakaran, R.; Renganathan, N. G.; Pitchumani, S. & Muniyanndi, N.- J. Power Sources, 89, p.80 (2000).
- 21. Choe, H. S.; Giaccami, J.; Alamgir, M, & Abraham, K. M. - Electrochim. Acta, 40, p.2289 (1995).
- 22. Song, J.Y.; Wang, Y. Y. & Wan C. C. J. Power Sources , 77, p. 183 (1999).
- 23. Appetecchi, G. B.; Hassoun, J.; Scrosati, B.; Croce, F.; Cassel, F. & Salomon, M. - J. Power Sources, 124, p.246 (2003).
- 24. Croce, F.; Persi, L.; Scrosati, B.; Serraino-Fiory, F.; Plichta, E. & Hendrickson, M. A. - Electrochim. Acta, 46, p.2457 (2001).
- 25. Morita, M.; Fujisaki, T.; Yoshimoto, N. & Ishikawa, M. - Electrochim. Acta, 46, p.1565 (2001).
- 26. Singhal. R. G.; Capracotta, M. D.; Martin, J. D.; Khan, S. A. & Fedkiw, P. S. - J. Power Sources, 128, p.247 (2004).
- 27. Bloise, A. C.; Tambelli, C. C.; Franco, R. W. A.; Donoso, J. P.; Magon, C. J.; Souza, M. F.; Rasario, A. V. & Pereira, E.C.- Electrochim Acta, 46, p.1571 (2001).
- 28. Cheng, C. L.; Wan, C. C.; Wang, Y. Y. & Wu, M. S. - J. Power Sources, In Press (2005).
- 29. Bruce P. G. & Vincent C. A. - J. Electroanal. Chem., 1, p.225 (1987).
- 30. Watanabe, M.; Sanui, K.; Ogata, N.; Kobayashi, T. & Ohiaki, Z. - J. Appl. Phys., 57, p.123 (1985).
- 31. Binod, K.; Lawrance, G. & Scanlon, G. - J. Power Sources, 52, p.261 (1994).
- 32. Croce, F.; Appetecchi, G. B.; Perci, L. & Scrosati B. - Nature, 394, p.456 (1998).
- 33. Saika, D. & Kumar, A. - Electrochem Acta, 49, p.2581 (2004).
- 34. Liu Y.; Lee J. Y & Hong L. - J. Power Sources 129 p.303 (2004).
- 35. Qian, X.; Gu, N.; Cheng, Z.; Yang, X.; Wang, E. & Dong, S. - Electrochim. Acta 46, p.1829 (2001).
- 36. Croce, F.; Persi, L.; Scrosati, B.; Serraino-Fiory, F.; Plichta, E. & Hendrikson, M. A. - Electrochim.Acta 46 p.2457 (2001).
- 37. Capuoano, F.; Croce, F. & Scrosati, B. - J. Electrochem. Soc., 138,p.1918 (1991).
- 38. Fauteax, D. - J. Electrochem Soc. Proc. 94-28, p.379 (1994).
- 39. Kuratomi, J.; Iguchi, T.; Bando, T.; Aihara, Y.; Ono, T. & Kuwana, K. - J. Power Sources 97-98, p.801 (2001).
- 40. Kanamura, K.; Tamura, H.; Shiraishi, S. & Takehara, Z. - J. Electrochem. Soc. 142, p.340 (1995).
- 41. Evans, J.; Vincent, C. A. & Bruce, P. G. - Polymer 28, p.2325 (1987).
- 42. Nasef, M. M. & Saidi, H. - Mater. Chem. Phys., 25, In press (2005).
Publication Dates
-
Publication in this collection
02 Oct 2006 -
Date of issue
June 2006
History
-
Received
18 Apr 2005 -
Reviewed
13 July 2005 -
Accepted
19 Aug 2005