Resumos
Inúmeros processos podem ser utilizados para produção de materiais poliméricos. Cada processo apresenta características peculiares, que permitem produzir resinas com as mais variadas propriedades, visando a diferentes aplicações do material polimérico final. Os processos de polimerização em suspensão são bastante empregados para produção de resinas poliméricas por apresentarem muitas vantagens, como a facilidade de separação, fácil remoção de calor e controle de temperatura e, principalmente, pelos baixos níveis de impureza e de aditivação no produto final. Por isso, processos de polimerização em suspensão são apropriados para obtenção de produtos para aplicações biotecnológicas e médicas, dentre outras. O objetivo principal deste trabalho é apresentar uma discussão dos aspectos fundamentais inerentes aos processos de polimerização em suspensão, visando a uma compreensão do efeito das principais variáveis de processo sobre o desempenho de polimerizações em suspensão. Mostra-se também que a espectroscopia do infravermelho próximo (NIRS) pode ser muito útil para permitir o monitoramento e o controle em tempo real de processos de polimerização em suspensão, para a obtenção de materiais poliméricos com características especiais.
Polimerização; suspensão; espectroscopia; infravermelho próximo
Several processes can be employed for manufacturing polymer materials. Each process presents some intrinsic features that lead to production of resins with peculiar properties, which define the end-use application of the final polymeric material. Suspension polymerization processes are extensively used because of their many advantages, such as the easy separation of the polymer particles, easy removal of the heat of reaction, easy temperature control and low levels of impurities and additives in the final polymer material. For this reason, suspension polymerization processes are suitable to produce polymer resins for biotechnological and medical applications. The main objective of this work is to discuss the fundamental aspects of suspension polymerization processes, focusing upon the effects of the main process variables on the performance of suspension polymerizations. Additionally, it is shown that near infrared spectroscopy (NIRS) can be useful for monitoring and real time control of suspension processes, allowing for production of polymer beads with controlled morphology.
Polymerization; suspension; spectroscopy; near infrared
ARTIGO DE REVISÃO
Uma revisão sobre os processos de polimerização em suspensão
A review on suspension polymerization processes
Fabricio Machado; Enrique L. Lima; José Carlos Pinto
Programa de Engenharia Química, COPPE, UFRJ
Autor para correspondência Autor para correspondência: José Carlos Pinto Programa de Engenharia Química, COPPE, UFRJ Cidade Universitária CP 68502 CEP 21945-970, Rio de Janeiro, RJ, Brasil E-mail: pinto@peq.coppe.ufrj.br
RESUMO
Inúmeros processos podem ser utilizados para produção de materiais poliméricos. Cada processo apresenta características peculiares, que permitem produzir resinas com as mais variadas propriedades, visando a diferentes aplicações do material polimérico final. Os processos de polimerização em suspensão são bastante empregados para produção de resinas poliméricas por apresentarem muitas vantagens, como a facilidade de separação, fácil remoção de calor e controle de temperatura e, principalmente, pelos baixos níveis de impureza e de aditivação no produto final. Por isso, processos de polimerização em suspensão são apropriados para obtenção de produtos para aplicações biotecnológicas e médicas, dentre outras. O objetivo principal deste trabalho é apresentar uma discussão dos aspectos fundamentais inerentes aos processos de polimerização em suspensão, visando a uma compreensão do efeito das principais variáveis de processo sobre o desempenho de polimerizações em suspensão. Mostra-se também que a espectroscopia do infravermelho próximo (NIRS) pode ser muito útil para permitir o monitoramento e o controle em tempo real de processos de polimerização em suspensão, para a obtenção de materiais poliméricos com características especiais.
Palavras-chave: Polimerização, suspensão, espectroscopia, infravermelho próximo.
ABSTRACT
Several processes can be employed for manufacturing polymer materials. Each process presents some intrinsic features that lead to production of resins with peculiar properties, which define the end-use application of the final polymeric material. Suspension polymerization processes are extensively used because of their many advantages, such as the easy separation of the polymer particles, easy removal of the heat of reaction, easy temperature control and low levels of impurities and additives in the final polymer material. For this reason, suspension polymerization processes are suitable to produce polymer resins for biotechnological and medical applications. The main objective of this work is to discuss the fundamental aspects of suspension polymerization processes, focusing upon the effects of the main process variables on the performance of suspension polymerizations. Additionally, it is shown that near infrared spectroscopy (NIRS) can be useful for monitoring and real time control of suspension processes, allowing for production of polymer beads with controlled morphology.
Keywords: Polymerization, suspension, spectroscopy, near infrared.
Introdução
Polímeros sintéticos podem ser obtidos pela combinação de monômeros através de um grande número de mecanismos de reação, incluindo polimerizações em cadeia (por exemplo, polimerizações via radicais livres, iônica, Ziegler-Natta) e em etapas (por exemplo, polimerizações funcionais). A arquitetura macromolecular (distribuição de massa molar, distribuição de composição do copolímero, distribuição de comprimento de blocos, distribuição de ramificações, estereorregularidade, etc.) depende não só da natureza química dos monômeros, do tipo de mecanismo de polimerização e estado físico do sistema reagente, mas também do tipo de processo e da configuração do reator[1].Para facilitar a manipulação do material polimérico final e a operação do processo de polimerização, processos heterogêneos de polimerização são empregados para a produção de polímero na forma de partículas (como os conhecidos processos de polimerização em emulsão, em dispersão, por precipitação e em suspensão)[2]. Cada processo apresenta características peculiares, que permitem produzir resinas com as mais variadas propriedades, visando a diferentes aplicações do material polimérico. Os processos de polimerização em suspensão são apropriados para obtenção de produtos para aplicações biotecnológicas e médicas, como por exemplo, partículas com morfologia controlada para uso em embolização vascular[3], como suporte para enzimas[4] ou como cimento ósseo para tratamento de osteoporose e em cirurgias dentárias[5,6].
Um típico sistema de polimerização em suspensão apresenta um ou mais monômeros insolúveis na água, contendo um iniciador solúvel na fase orgânica. Essas espécies são dispersas numa fase aquosa contínua por uma combinação de forte agitação e uso de pequenas quantidades de agentes de suspensão (estabilizantes). Uma condição satisfatória de agitação mecânica é suficiente para manter o monômero na forma de gotículas, que são lentamente convertidas de um estado líquido de alta mobilidade para uma mistura altamente viscosa (conversão em torno de 20 a 60%) e, finalmente, para uma partícula de polímero dura. Os estabilizantes impedem a coalescência das gotas orgânicas suspensas na fase aquosa, estabilizando a gota de polímero. A tendência de aglomeração das gotas pode ficar crítica, quando a polimerização avança para o ponto onde a gota de polímero torna-se pegajosa[7].Um dos desafios da técnica de polimerização em suspensão é prevenir a aglomeração da fase orgânica.
A técnica de polimerização em suspensão procura reunir as vantagens dos processos de polimerização em massa, solução e emulsão, ao mesmo tempo em que procura eliminar algumas de suas desvantagens. A polimerização em suspensão corresponde grosseiramente a uma polimerização em massa dentro de cada gotícula de monômero suspensa no meio aquoso. Dessa forma, aumenta-se a concentração do monômero e, conseqüentemente, as velocidades de reação (quando comparada à polimerização em solução). Como as partículas formadas são relativamente grandes (tipicamente na faixa de 20 a 500 mm) e como a separação do produto final por sedimentação é fácil, não é necessário adicionar emulsificantes e nem agentes que previnam e/ou promovam a coagulação, de maneira que os níveis de impureza e aditivação no produto final são usualmente muito baixos, quando comparado com a polimerização em emulsão. O produto final é obtido na forma de partículas esféricas de tamanho e forma relativamente homogêneos. Além disso, o tamanho médio da partícula e a distribuição de tamanhos das partículas podem ser controlados de forma simples numa faixa relativamente estreita, manipulando-se a velocidade de agitação e a quantidade de agente de suspensão. Devido à faixa de tamanho, a recuperação final das partículas numa corrente aquosa ou gasosa é relativamente fácil, com baixo custo de separação, quando comparado com a polimerização em emulsão, em solução e em massa. A baixa viscosidade da lama mesmo a altas conversões facilita a operação do sistema e permite a obtenção de suspensões bastante homogêneas. A baixa viscosidade também contribui com a fácil remoção de calor e com o controle eficiente da temperatura. Além disso, o meio de suspensão absorve parte do calor produzido, reduzindo o impacto térmico sobre a unidade. Por todas essas razões, observa-se que um grande número de resinas comerciais importantes é produzido por polimerização em suspensão. Dentre esses, os exemplos comerciais mais importantes são os da produção de poli(cloreto de vinila) e copolímeros, de poliestireno e copolímeros, de poli(acetato de vinila) e copolímeros e de poli(metacrilato de metila) e copolímeros.
Processos de Polimerização em Suspensão
Sabe-se que a natureza do processo de polimerização pode afetar o comportamento cinético, a microestrutura, a arquitetura molecular e a homogeneidade do produto formado[8]. Por isso é importante definir de maneira apropriada o processo de polimerização em suspensão empregado. Os principais processos de polimerização em suspensão usados comercialmente podem ser divididos em sete tipos, descritos a seguir.
Polimerização em suspensão do tipo pérola
O monômero funciona como solvente do polímero produzido. As gotas de monômero passam de um estado de elevada viscosidade até transformam-se em pequenas esferas de material sólido[7]. O caso típico é o da polimerização do estireno em batelada. A distribuição final de tamanhos das pérolas de polímero é geralmente o resultado da distribuição do tamanho das gotas da dispersão inicial de monômero em água e de um processo de aglomeração controlada no primeiro estágio da polimerização. Mais especificamente, a suspensão sofre as transições descritas a seguir[1]. 1) durante o período inicial a distribuição dos tamanhos de gotas é geralmente estreita e se comporta como uma suspensão de um líquido em outro líquido; 2) durante o período intermediário ocorre a formação de uma mistura pegajosa, caracterizada pelo aumento expressivo da viscosidade da fase dispersa e pela correspondente redução da velocidade de quebra. A distribuição de tamanhos das partículas se alarga e o controle de aglomeração se torna crítico. O equilíbrio que se estabelece entre as taxas de coalescência e quebramento define o grau de estabilidade da suspensão; e 3) durante o último estágio, se a suspensão for instável, ocorre a coalescência descontrolada. Caso contrário, a coalescência cessa e a partícula adquire a sua forma rígida final. O limite crítico que separa o segundo intervalo do terceiro é chamado de ponto de identificação da partícula (PIP), porque deixa de ser possível o controle da distribuição de tamanhos de partícula a partir desse ponto. O PIP depende das condições de operação, mas corresponde quase sempre a uma conversão na faixa de 65 a 80% do monômero.
Polimerização em suspensão do tipo granular
Nesse caso, o polímero não é dissolvido pelo seu monômero. A polimerização ocorre em cada gota de monômero, mas o polímero formado precipita ao longo da polimerização. A evolução da viscosidade da fase dispersa durante os primeiros estágios da polimerização é, portanto, mais lenta, já que a fase dispersa contém monômero livre. No final são obtidos pós ou grãos opacos e irregulares, que apresentam freqüentemente a forma de cachos de uva (aglomerados de grãos primários). O caso típico é o da polimerização do cloreto de vinila em batelada.
Polimerização em massa-suspensão ou semi-suspensão
Consiste num processo de polimerização em suspensão conduzido em duas fases. Na primeira fase é feita uma polimerização em massa. Quando a conversão alcança um determinado valor (por exemplo, 25-30%), a mistura reacional altamente viscosa é transferida para um reator que contém água e estabilizante, onde a polimerização prossegue até que se alcance a conversão desejada[7]. A operação pode ser conduzida em modo contínuo ou em batelada. A operação em semi-suspensão permite que curvas de distribuição de tamanho de partícula mais estreitas sejam obtidas. Acredita-se que esse efeito seja devido à redução do segundo estágio da polimerização em suspensão.
Polimerização Inversa
O uso de polimerização em suspensão inversa é um desenvolvimento relativamente novo. Polimerização em suspensão inversa é conduzida com a dispersão de monômeros solúveis em água numa matriz orgânica contínua. Termodinamicamente, a dispersão é instável e requer contínua agitação e adição de agentes estabilizantes. A iniciação geralmente é feita termicamente ou quimicamente, com um radical livre de um azocomposto ou percomposto[2,9,10]. No caso do uso de um único componente iniciador, a polimerização pode ser iniciada pela decomposição do iniciador na fase orgânica, na fase aquosa ou em ambas as fases, dependendo da partição do iniciador nas duas fases. Quando é usado um par redox para iniciação, ao menos um dos componentes tem que ser segregado do monômero, para prevenir a polimerização antes da dispersão inversa ser estabelecida. O oxidante geralmente entra com o monômero na dispersão aquosa inversa inicial. O agente redutor é introduzido depois como uma solução aquosa, para começar a polimerização. Alternativamente, ambos os agentes oxidantes e redutores em solução aquosa podem ser introduzidos separadamente na dispersão aquosa do monômero na fase orgânica agitada[11].
Polimerização em suspensão-emulsão
Esse processo foi desenvolvido recentemente e combina diferentes processos heterogêneos de polimerização, explorando as vantagens dos processos de polimerização em suspensão e emulsão para produzir partículas com morfologia casca-núcleo e/ou distribuição de massas molares bimodais[4]. No primeiro caso, um núcleo rígido é formado pelas partículas obtidas pela polimerização em suspensão, enquanto uma casca porosa é formada pelas partículas resultantes do processo em emulsão. Essas partículas podem ser usadas como suporte de enzimas em aplicações biotecnológicas[12,13]. A Figura 1 ilustra uma partícula tipicamente obtida pelo processo de polimerização em suspensão-emulsão[4]. No segundo caso, material de baixa massa molar é formado em suspensão, enquanto um material de massa molar mais alta resulta do processo em emulsão. O processo permite ainda a produção de compósitos e blendas poliméricas in situ durante copolimerizações de diferentes monômeros[14].
Polimerização em dispersão
Difere dos processos de polimerização em suspensão clássicos por se tratar de um sistema de polimerização inicialmente homogêneo (composto pelo monômero, um solvente orgânico, o iniciador solúvel na fase orgânica e um agente estabilizante). O sistema torna-se heterogêneo pelo fato do polímero formado durante a polimerização ser insolúvel no solvente. As partículas poliméricas formadas são estabilizadas graças à ação do agente estabilizante da suspensão. A polimerização prossegue no meio contínuo e nas partículas poliméricas, que absorvem monômeros e radicais presentes na fase contínua. Processos de polimerização em dispersão geralmente conduzem a partículas com tamanhos intermediários entre aqueles obtidos via processos em emulsão e suspensão clássicos, na faixa de 1 a 10 mm de diâmetro[15]. A seleção do solvente orgânico depende sobretudo de sua miscibilidade com os outros constituintes do sistema reacional. O ideal é que o monômero, o agente estabilizante e o iniciador da polimerização apresentem completa solubilidade no solvente, enquanto que o polímero seja insolúvel no solvente e precipite durante a polimerização[16]. Tipicamente, álcoois como o etanol e o metanol, em diferentes concentrações, são solventes apropriados para condução de polimerizações de monômeros como o estireno, o acrilato de butila, o metacrilato de metila, etc[16-18].
Polimerização em microssuspensão
Este processo é apropriado para formação de partículas com tamanhos intermediários às partículas produzidas por reações de polimerização em suspensão e emulsão clássicas. Este processo apresenta características cinéticas essencialmente semelhantes às de um processo em suspensão (cada gotícula de monômero age como um micro-reator em massa), porém atingindo conversões similares às do processo em emulsão. Por conduzir a conversão quase completa (em tono de 100%), este processo é mais econômico, eliminando etapas de processamento pós-reação necessárias para remoção e recuperação de monômero residual[19]. Um processo de polimerização em dispersão ideal deve se comportar como uma polimerização em suspensão clássica. As gotas de monômero são geralmente estabilizadas por estabilizantes polares, entretanto o iniciador da polimerização é solúvel na fase orgânica. Devido à baixa tensão interfacial, o diâmetro das gotas de monômero pode ser reduzido a aproximadamente 0,1 a 10 mm, por meio de homogeneização mecânica, com a aplicação de elevadas taxas de cisalhamento. Neste processo, a polimerização também pode ocorrer em emulsão na fase aquosa, contudo em pequena extensão, mesmo que o monômero possua reduzida solubilidade na água[19,20]. As partículas formadas em emulsão podem apresentar propriedades completamente diferentes das obtidas pelo processo em suspensão; por isso, o peso molecular, a composição e o tamanho de partícula podem exercer efeito significativo sobre a propriedades finais, influenciando o desempenho do material polimérico final[19].
Estabilizantes: Agentes de Suspensão
A polimerização em suspensão geralmente requer a adição de pequenas quantidades de um estabilizante para impedir a coalescência e a quebra de gotas durante a polimerização. O estabilizante afeta o tamanho e a forma das partículas, bem como a cristalinidade e a transparência finais das pérolas. Além disso, as vantagens normalmente associadas à facilidade de remoção de calor e às baixas viscosidades são apenas atingidas de fato se a agitação for capaz de manter as partículas em suspensão durante todo o curso da polimerização, o que pode ser facilitado se um agente de suspensão é adicionado para garantir a ausência de coalescência[21].
O estabilizante típico é constituído por uma mistura de polímeros polares, com caráter simultaneamente hidrofílico e hidrofóbico, embora estabilizantes inorgânicos insolúveis em ambas as fases possam também ser usados. O estabilizante polimérico, quando dissolvido na fase aquosa, pode atuar de duas formas. Em primeiro lugar, ele diminui a tensão interfacial entre as gotas de monômero e a água, promovendo a dispersão das gotas. Em segundo lugar, as moléculas estabilizantes são adsorvidas sobre a superfície das gotas de monômero, produzindo uma fina camada que evita a coalescência das gotas quando ocorre a colisão entre elas[7]. A tensão interfacial entre dois líquidos imiscíveis depende da temperatura, da concentração e da natureza química (por exemplo, grupamento hidroxila) dos agentes estabilizadores presentes na superfície da gota[22], de maneira que a eficiência dos agentes de suspensão depende das condições de operação.
Um dos mais importantes fenômenos que governam a estabilidade da suspensão é a adsorção do estabilizante sobre a superfície da fase dispersa. Lazrak et al.[23] observaram que, quando a velocidade de agitação é aumentada, o diâmetro médio das gotas diminui sob a ação da tensão de cisalhamento. Então, para uma mesma quantidade de monômero, a área interfacial é aumentada, havendo conseqüentemente a necessidade de uma quantidade maior de estabilizante para cobrir toda a superfície da gota. Quando a agitação é muito alta, pode ocorrer também a dessorção do agente de suspensão, resultando na redução da espessura da camada de proteção e alterando a eficiência do estabilizante. Dessa forma, uma agitação excessiva pode acabar contribuindo com ocorrência de aglomeração das partículas.
O poli(álcool vinílico) (PVA) é um dos estabilizantes mais utilizados em polimerizações em suspensão; entretanto, muitos outros estabilizantes podem também ser empregados na polimerização em suspensão, como, por exemplo, o poliestireno sulfonado (PES)[24-26], o trifosfato de cálcio (TCP)[27,28], o hidroxietilcelulose (HEC)[27],o poli(metacrilato de amônio) (APMA)[29], o 2-etil hexil sulfato de sódio[30], o dodecil benzeno sulfonato de sódio (SDBS)[28] e a gelatina[31], dentre outros. O PVA é comercialmente caracterizado por duas propriedades: a viscosidade da solução aquosa, que está diretamente ligada a sua massa molar; e o grau de hidrólise, que se refere à percentagem de radicais acetil que foram substituídos por radicais hidroxila durante a reação de hidrólise do poli(acetato de vinila). A variação dessas duas propriedades permite obter uma ampla variedade de resinas de PVA com propriedades específicas. Sabe-se que o aumento da massa molar e do grau de hidrólise gera um produto com melhores propriedades mecânicas, porém menos solúvel em água. Quando o PVA é utilizado como colóide protetor, a sua eficiência na estabilização do sistema depende da sua massa molar, do grau de hidrólise, da freqüência de ramificações de cadeias longas e curtas e da distribuição dos grupos acetil na cadeia[32].
A maioria dos estabilizantes utilizados em polimerizações em suspensão é pouco ou totalmente solúvel em água. Usualmente são também utilizadas partículas inorgânicas como estabilizantes, que adsorvem na interface água/óleo. Surfactantes às vezes são também adicionados em baixas concentrações, com a finalidade de ajudar o processo de dispersão inicial. Ao contrário de estabilizantes poliméricos, muitos tipos de estabilizantes inorgânicos podem ser removidos após a reação de polimerização por lavagem com um ácido, uma base ou por meio de abrasão mecânica da superfície. Além disso, partículas inorgânicas são geralmente mais baratas que estabilizantes poliméricos[2].
Eletrólitos também são usados como complementos dos surfactantes. Tampões e outros eletrólitos usualmente aumentam a tensão superficial entre as fases aquosa e monomérica. Eles também reduzem a solubilidade dos monômeros na fase aquosa e freqüentemente resultam na diminuição da partícula. Aditivos podem ser utilizados para aumentar a viscosidade da fase aquosa e, conseqüentemente, reduzir o número de colisões entre as gotas, responsáveis pela coalescência ou aglomeração[33].
Iniciadores
Os iniciadores utilizados em polimerizações em suspensão são solúveis na fase orgânica, já que a polimerização ocorre nas gotas do monômero. Tipicamente a composição do iniciador fica na faixa de 0.1 a 0.5% em peso do monômero. A alimentação freqüentemente é feita antes da suspensão alcançar a temperatura de reação, na forma de uma solução de monômero. Como a transferência de massa pode constituir um problema relevante para essa tecnologia, a carga de iniciador é normalmente alimentada no início do processo.
Durante a escolha do iniciador, o critério de solubilidade em monômero é determinante. Usualmente em polimerizações em suspensão são utilizados como iniciadores os peróxidos orgânicos e azocompostos, pois estes quase sempre são solúveis na fase monomérica orgânica.
Iniciação
Consiste na etapa responsável pela formação de radicais livres numa reação de polimerização em cadeia. Os iniciadores usualmente sofrem homólise, gerando os radicais livres por decomposição térmica ou fotoquímica (luz ultravioleta). Nessa classe de iniciadores, os mais comuns são o peróxido de benzoíla (BPO) e o azobis(isobutironitrila) (AIBN). Embora os peróxidos sejam mais utilizados como iniciadores por causa dos preços mais vantajosos, os azocompostos são preferidos quando o objetivo é a obtenção de materiais com baixo grau de ramificação[34].
A etapa de iniciação é composta por duas reações elementares: 1) geração do radical primário de iniciador via decomposição térmica; e 2) combinação do radical primário com uma molécula de monômero, resultando na formação da cadeia polimérica composta de uma unidade monomérica.

A velocidade de decomposição do iniciador geralmente segue uma cinética de primeira ordem, na forma:

A forma com que a Equação 1 está escrita indica que todos os radicais gerados pela decomposição térmica das moléculas de iniciador são usados para formar o primeiro radical polimérico P1. Entretanto, isso não acontece na prática, pois apenas parte dos radicais é aproveitada de forma efetiva. Para corrigir este problema, é preciso incluir na equação da taxa de formação de radicais poliméricos o parâmetro f, que quantifica a eficiência do iniciador, informando que fração dos radicais primários de iniciador gerados realmente será utilizada para formação dos radicais poliméricos. A taxa de geração dos radicais poliméricos pode ser escrita como:

O desperdício dos radicais poliméricos ocorre devido ao efeito gaiola (cage effect), que resulta do confinamento dos radicais pelas moléculas do solvente, do monômero ou do polímero. Assim, quanto maior a resistência que o meio oferece à difusão dos radicais formados, maior a probabilidade de que estes venham a se recombinar ou participar de reações paralelas que não resultem na polimerização. A eficiência e o tempo de meia vida do iniciador dependem da natureza física do meio em que o iniciador se decompõe, da concentração de polímero no meio reacional, da temperatura, da mobilidade do radical primário, da massa molar das espécies e da composição do meio[35,36]. A Tabela 1 ilustra o tempo de meia-vida de peróxidos orgânicos comuns, como função da temperatura de decomposição[15,37].
Wolff e René Bos[35] descreveram detalhadamente o efeito gaiola. As várias espécies moleculares presentes no meio reacional tendem a formar uma célula que inibe a difusão dos radicais para fora dela. Como resultado desse fenômeno, apenas uma fração dos radicais gerados consegue escapar para iniciar novas cadeias poliméricas, com velocidade que depende do seu coeficiente de difusão (DI). A difusão do radical primário aprisionado pode ser compreendida com um modelo geométrico formado por duas esferas concêntricas de raios r1 e r2. Admite-se que os radicais primários são gerados unicamente na esfera de reação de iniciador de raio r1, localizada dentro de uma grande esfera de difusão de raio r2. Apenas os radicais que escapam da esfera de raio r2 podem reagir com o monômero para formar novas cadeia poliméricas. Este mecanismo pode ser descrito pela seguinte equação.

onde kio é a constante cinética para a reação de iniciação; f é a eficiência do iniciador; f0 é a eficiência do iniciador na ausência de impedimentos difusionais; e [M] é a concentração de monômero.
Freqüentemente, a eficiência do iniciador é considerada constante. Entretanto, no regime de alta viscosidade do meio reacional, a eficiência do iniciador pode diminuir de forma significativa. Dubé et al.[38] propuseram uma equação semi-empírica que descreve a mudança na eficiência de iniciador para polimerizações conduzidas em massa ou solução, quando o volume livre da mistura reacional torna-se menor que um certo volume livre crítico do sistema reacional

onde f0 é a eficiência do iniciador na ausência de impedimentos difusionais, C é um parâmetro ajustável que modifica a variação de mudança da eficiência; vf é o volume livre do meio reacional, e vfcrift é o volume livre crítico do meio reacional, que depende da temperatura e do tipo de iniciador.
Fenômeno de Quebra e Coalescência
Nos processos de polimerização em suspensão a descrição e compreensão dos fenômenos de quebra e coalescência das gotas são de grande importância, por causa da influência que tais mecanismos exercem principalmente sobre o tamanho e a distribuição do tamanho das partículas.
Muitos processos na engenharia envolvem misturas de dois líquidos imiscíveis na forma de uma dispersão. Em geral, nos processos que envolvem o fenômeno da dispersão, é desejável melhorar o mecanismo de transferência de massa através do aumento da área interfacial. A área de transferência de massa é determinada pelo tamanho da gota e pela fração volumétrica da fase dispersa. A distribuição do tamanho da gota obtida em uma dispersão é resultado dos mecanismos de quebra e coalescência que ocorrem simultaneamente[39,40].
A polimerização em suspensão geralmente requer a adição de pequenas quantidades de um estabilizante, para impedir a coalescência e a quebra de gotas durante a polimerização. A distribuição de tamanhos de partícula e, conseqüentemente, o tamanho das pérolas de polímero formadas dependem do balanço entre as taxas de quebra e coalescência das gotas. Este mecanismo pode ser controlado pela velocidade de agitação da mistura reacional, pela forma do impelidor, pela fração volumétrica da fase monomérica e pela concentração e a massa molar do estabilizador usado[41-44]. A tensão interfacial, o grau de agitação e o planejamento do sistema reator/agitação governam a dispersão das gotas de monômero, tipicamente com diâmetros na faixa de 20 à 500 µm[2,45].
Em contraste com a polimerização em emulsão, a velocidade de agitação na polimerização em suspensão determina a distribuição do tamanho das partículas. A formação de uma dispersão de gotas estável requer a combinação de uma série de ações estabilizantes (agitação, agentes de suspensão, ajuste da densidade e/ou viscosidade das fases). O tamanho da partícula é controlado por agitação e pela adição de estabilizadores e/ou surfactantes[27,46]. A distribuição de tamanho de partícula (DTP) num processo em suspensão é determinante para estabilidade da mistura polimérica. Quando empregadas em processos cromatográficos e na manufatura de suportes, as partículas obtidas de processos de polimerização em suspensão devem apresentar DTP estreita.
Durante uma polimerização em suspensão clássica, o processo de formação das partículas é geralmente dividido em três estágios, como descrito anteriormente[47]. Alguns trabalhos são dedicados exclusivamente à descrição da evolução da DTP e à modelagem do mecanismo dinâmico da quebra e coalescência em sistemas de polimerização em suspensão durante os diferentes estágios da reação[22,39,46-50].
Em processos de polimerização em suspensão em batelada os fenômenos de quebramento e coalescência são os mais importantes. Assim, o balanço populacional utilizado para descrever a evolução da DTP pode ser dado na forma:

onde os dois primeiros termos após a igualdade caracterizam o desaparecimento e o aparecimento de partículas de massa m devido à coalescência. Os dois últimos termos após a igualdade representam o aparecimento e desaparecimento de partículas de massa m devido ao quebramento. NP é o número total de partículas, Kc (m, m) é a constante de taxa de coalescência, b (m, m) é probabilidade de formação de novas gotas, g (m) é a constante de probabilidade de quebra, l (m) é o número médio de gotas novas geradas pela quebra de uma gota de massa m, f (m) é a função de densidade da distribuição de tamanho de partículas de polímero.
A Equação 6, quando integrada para todas as partículas de todos os tamanhos, resulta em:

onde o primeiro termo após a igualdade refere-se ao desaparecimento de partículas após a coalescência e o segundo termo após a igualdade refere-se ao aparecimento de partículas devido ao quebramento. Métodos numéricos baseados em aproximações polinomiais foram desenvolvidos para permitir a solução eficiente das equações de balanço populacional que descrevem a polimerização em suspensão[47].
Efeitos Gel e Vítreo
Os efeitos gel e vítreo são fenômenos cinéticos que aparecem durante o decorrer da reação de polimerização. O efeito gel surge tipicamente quando as conversões de monômero atingem a faixa de 20 a 40% e está relacionado ao aumento de viscosidade no meio reacional. A mobilidade das cadeias de crescimento é fortemente reduzida pela alta viscosidade, de forma que a velocidade de terminação cai drasticamente[21], resultando numa auto-aceleração da polimerização. Este fenômeno afeta fortemente as propriedades finais do polímero e conduz à produção de resinas com larga distribuição de massas molares. Além disso, pode causar também a elevação brusca de temperatura no reator. A presença do efeito gel introduz não-linearidades relevantes no modelo de polimerização via radicais livres[1,51]. De acordo com Crowley e Choi[52], essas não linearidades são acentuadas quando o aumento da viscosidade também diminui o coeficiente de transferência de calor efetivo entre as paredes do reator e o fluido refrigerante na jaqueta. Quando isso ocorre, o polímero preso nas paredes do reator causa a diminuição significativa da capacidade de remoção de calor do reator, resultando finalmente em descontrole térmico da operação.
Designa-se usualmente como efeito vítreo à diminuição da constante cinética de propagação causada pela diminuição da mobilidade das moléculas de monômero num meio altamente viscoso, causando conseqüente redução das velocidades de reação e das massas molares médias das cadeias formadas. Como no caso anterior, estes fenômenos conduzem ao alargamento da distribuição de massas molares e afetam fortemente as propriedades finais dos polímeros[35,53,54]. O efeito vítreo aparece em polimerizações em que a temperatura de reação se encontra abaixo da temperatura de transição vítrea do polímero, em conversões superiores a 90%. A conseqüência deste fenômeno é o "congelamento" da mistura reacional. Para a conversão limite, a temperatura de transição vítrea da mistura polímero/monômero torna-se igual à temperatura de polimerização[1].
Em processos de polimerização via radicais livres, todas as etapas de reação podem ser bastante afetadas por limitações difusionais, já que esses mecanismos envolvem reações entre moléculas de comprimento elevado. Portanto, o aumento da viscosidade do meio causado pelo aumento da concentração de polímero pode afetar bastante as velocidades elementares de reação, que em última análise definem a velocidade da reação e a estrutura molecular do material final[38,55-58].
Correlações para os efeitos gel e vítreo
As mudanças nas constantes de terminação (kT) e propagação (kP), devidas às limitações impostas pelo aumento da viscosidade do meio reacional, geralmente são expressas como funções da temperatura e da composição do meio, embora possam também ser representadas como funções da viscosidade do meio[59,60] ou por outras formas empíricas[17,61,62]. Correlações baseadas na teoria do volume livre são também bastante utilizadas, pois o volume livre pode caracterizar extremamente bem as resistências à difusão em meios complexos[54,63].
O volume livre pode ser definido de forma simples como o espaço vazio existente no meio, que não é ocupado pelas moléculas. Quando a mistura polimérica se aproxima do ponto de transição vítrea, o volume livre diminui, até que o movimento de translação molecular cessa[64]. Conseqüentemente, nesse momento a velocidade de reação cai a zero. A fração de volume livre do sistema é afetada pela temperatura, viscosidade e densidade do meio reacional.
ONeil et al.[65,66] examinaram experimentalmente a consistência da teoria do volume livre, quando aplicada para descrever o efeito gel em sistemas de polimerização via radical livre. Os autores mostraram que a teoria do volume livre prediz com sucesso o efeito gel, e que a diminuição do volume livre pode ser relacionada com o aumento da viscosidade do meio reacional, à medida que o monômero é convertido a polímero.
Influência da Solubilidade do Monômero na Fase Aquosa
Nas polimerizações em suspensão, se os monômeros envolvidos são insolúveis em água, o comportamento cinético e as propriedades do polímero são essencialmente os mesmos daqueles obtidos na polimerização em massa. Porém, se os monômeros têm solubilidade bastante diferentes em água, podem ocorrer mudanças significativas da cinética de polimerização e das propriedades do polímero final. Existem duas formas de reduzir a solubilidade de monômeros em água em polimerizações em suspensão: tentar aumentar a transferência de monômero para a gota e/ou reduzir a solubilidade em água com a adição de aditivos (em geral, eletrólitos).
Para reduzir a solubilidade do monômero em água, normalmente adiciona-se sal à fase aquosa. Como o sal tem muito mais afinidade com a água que com o monômero orgânico, as variações de solubilidade são muito sensíveis à adição de pequenas quantidades de sal. Deve-se salientar, no entanto, que o sal também diminui a eficácia do agente estabilizante, de maneira que, para prevenir a coagulação da partícula, é necessária uma quantidade maior de estabilizante. O sal também contribui com o aumento da conversão do monômero, pois reduz a quantidade de monômero dissolvido na fase aquosa, e altera as propriedades do polímero, especialmente a distribuição de massas molares e de composição, por modificar a composição do meio[67]. Além disso, é uma prática comum nos processos industriais a adição de pequenas quantidades de sal para ajuste do pH, da densidade e da viscosidade da fase contínua, e também para prevenir a incrustação de polímero nas paredes do reator[68].
A adição de sal é eficiente mesmo em sistemas de polimerização que apresentem monômeros com elevada solubilidade na água. Machado et al.[68-70] avaliaram a influência de NaCl sobre o coeficiente de partição do ácido acrílico em copolimerizações em suspensão de acetato de vinila/ácido acrílico. Observou-se que o NaCl pode ser utilizado para aumentar o coeficiente de partição do acido acrílico entre as fases orgânica e aquosa. Contudo, é muito importante enfatizar que concentrações elevadas de sal podem conduzir à diminuição da estabilidade da suspensão (Figura 2), causando uma aglomeração das partículas de polímero, caso a agitação e a concentração de agente de suspensão permaneçam constante.
Influência do Uso de Solventes na Fase Orgânica
Solventes podem ser adicionados à fase orgânica para reduzir as velocidades de polimerização, reduzir as massas molares médias, prevenir o descontrole térmico resultante do efeito gel e/ou alterar a morfologia das partículas produzidas. Os teores de solvente devem ser baixos, no entanto, para prevenir a aglomeração, já que o aumento arbitrário do teor de solventes pode impedir que se atinja o PIP. Os solventes permitem que se obtenham estruturas porosas, já que as pérolas de polímero obtidas através do processo em suspensão são muito pouco porosas, apresentando área específica inferior a 0.1 m2/g. O uso de solventes expansíveis (por exemplo, pentano em estireno, acrilonitrila e metacrilonitrila) pode permitir a produção de espuma, fazendo-se a rápida descompressão do ambiente de reação[30,71,72]. Essa técnica é usada freqüentemente para a produção de espumas de poliestireno[71].
Em polimerizações via radicais livres as interações polares entre as moléculas de monômero e solvente durante as reações de propagação podem afetar as razões de reatividade. Quando as interações do tipo polar são importantes na estrutura de transição da reação de propagação, a polaridade do solvente pode afetar a taxa de propagação, a seletividade do radical e a reatividade do radical. Em copolimerizações é provável que o efeito polar seja mais importante nas propagações cruzadas, por causa das diferentes propriedades eletrônicas. Em homopolimerizações o efeito polar é menos importante. Estudos mostram que as razões de reatividade são sensíveis à natureza do solvente, embora não existam muitos dados disponíveis sobre correlações entre a reatividade dos monômeros e a constante dielétrica dos solventes. Os exemplos mais comuns de sensibilidade à natureza do solvente são de copolimerizações de estireno, tendo como comonômero ácido acrílico, acrilamida ou anidrido maléico[73].
Copolimerização
A copolimerização de espécies químicas distintas tem sido um processo extensamente empregado para modificar as propriedades dos polímeros. Escolhendo-se apropriadamente os comonômeros, as propriedades originais do polímero podem ser melhoradas e modificadas drasticamente, permitindo a ampliação da qualidade de materiais distintos que podem ser produzidos. Quando duas espécies monoméricas são usadas para obter um polímero, suas unidades méricas podem estar distribuídas ao longo de uma cadeia polimérica de várias maneiras: aleatoriamente segundo uma lei estocástica de distribuição (copolímeros estatísticos ou aleatórios), arranjados alternadamente ao longo da cadeia (copolímeros alternados), combinados em blocos distintos (copolímeros em bloco) ou apensados na forma de enxerto (copolímeros enxertados).
Quando todas as moléculas do copolímero têm exatamente o mesmo número de unidades estruturais, todas elas têm o mesmo grau de polimerização, a mesma massa molar e a mesma composição. Devido à natureza estatística dos processos de polimerização, é praticamente impossível obter moléculas de copolímero que contenham precisamente igual comprimento de cadeia e composição[74]. Por isso, o material resultante da copolimerização necessariamente apresenta uma certa distribuição característica de massas molares e composição. Essas distribuições em geral são acopladas e definem as propriedades e aplicações do produto final. Em processos de copolimerização, no entanto, a composição do copolímero e as seqüências de blocos de cada tipo de monômero ao longo da cadeia são consideradas as propriedades de mais elevada importância, pois definem em última instância a qualidade do produto polimérico final.
A copolimerização em suspensão pode ser feita mesmo que um dos monômeros tenha considerável solubilidade em água. Nesta situação, o monômero solúvel em água difunde para a fase orgânica, onde ocorre a copolimerização. Contudo, as copolimerizações conduzidas em suspensão são usualmente realizadas em batelada. Isso ocorre porque há resistências à transferência de massa e porque não é possível garantir que a alimentação de monômero ao longo da reação vá se distribuir uniformemente por todas as partículas previamente existentes. Isso significa que desvios significativos de composição podem ocorrer ao longo do tempo, resultando em materiais de composição heterogênea, o que não é costumeiramente desejado. Por isso, copolimerizações em suspensão são normalmente realizadas apenas nas condições azeotrópicas de reação (as composições do meio reacional e do copolímero produzido são iguais). O exemplo clássico é o da polimerização do estireno e da acrilonitrila, para produção de resina SAN[75-78]. Deve-se observar que as razões de reatividade em polimerizações em suspensão diferem das observadas nos processos em massa ou solução, possivelmente devido às diferenças de solubilidade dos monômeros na fase aquosa e às mudanças de coeficientes de atividade observadas em meios complexos[7].
Mecanismo de copolimerização
O tratamento quantitativo de copolimerizações em cadeia, incluindo aquelas conduzidas em processos de polimerização em suspensão, baseia-se na consideração de que a reatividade das cadeias em crescimento depende exclusivamente da atividade da unidade terminal (modelo último). Quando dois monômeros M1 e M2 são copolimerizados, existem quatro possíveis reações de propagação[79,80].

A partir do esquema indicado pela Equação 8 é possível encontrar uma equação (denominada de Equação de Mayo-Lewis) que relaciona a composição do copolímero com a composição dos monômeros no meio reacional, válida para conversões inferiores a 10%[81].

onde r1 e r2 são as razões de reatividade dos monômeros M1 e M2, definidas como r1 =  e r2 =
e r2 =  .
.
Existem alguns sistemas que não seguem a equação de Mayo-Lewis, de maneira que o modelo terminal pode não descrever bem a cinética de propagação. Para estes casos, utiliza-se o modelo penúltimo, que considera também o efeito da penúltima unidade mérica sobre a reatividade da cadeia polimérica. Dois tipos de modelo penúltimo já foram propostos: o modelo terminal explícito admite que as unidades terminal e penúltima do radical podem afetar a reatividade e a seletividade; e o modelo terminal implícito considera que apesar das unidades terminal e penúltima do radical polimérico afetarem a reatividade, apenas a unidade terminal afeta a seletividade[73]. O modelo penúltimo pode ser justificado de forma rigorosa pelo fato de que a espécie penúltima pode modificar significativamente tanto a mobilidade quanto o ambiente eletrônico do centro ativo da polimerização.
O mecanismo representado pelo modelo penúltimo é composto por quatro radicais distintos que podem combinar com os monômeros, gerando oito etapas distintas de propagação, como mostra a Equação 10. A introdução de etapas adicionais de propagação e o conseqüente aumento do numero de parâmetros cinéticos é a grande limitação prática do uso do modelo penúltimo em modelos de polimerização.

Razão de Reatividade
Existem vários métodos propostos para a determinação das razões de reatividade em sistemas de polimerização. Alguns métodos são gráficos, enquanto outros utilizam ferramentas de estimação de parâmetros e planejamento experimental. A aplicação destes métodos requer medidas da composição do copolímero e da concentração dos monômeros no meio reacional no decorrer da reação. Alguns destes métodos serão descritos a seguir.
Modelo terminal
A Equação 9, além de descrever a taxa de incorporação de M1 em relação à de M2, fornece também a composição instantânea do copolímero formado. A Equação de Mayo-Lewis usualmente é escrita como.

onde F1 é a fração molar de M1 no copolímero e fi a fração molar de Mi na mistura.
Os primeiros métodos gráficos propostos foram baseados em simplificações da Equação 9 e são difíceis e trabalhosos para serem implementados. Uma forma linear dessa equação[81] permite a determinação das constantes por regressão linear, sendo utilizado até hoje, para a determinação da razão de reatividade de pares monoméricos. Definindo-se F = m1/m2 e f = M1/M2 chega-se à Equação 12, que pode ser colocada na forma linear de diferentes maneiras, com a finalidade de facilitar o cálculo das razões de reatividade.

Outro método linear, embora avaliado somente por relações numéricas, é conhecido como o método YBR[82]. Técnicas de estimação de parâmetros baseadas no método dos mínimos quadrados são bastante utilizadas para o cálculo das razões de reatividade. Bons resultados são obtidos quando o planejamento experimental é utilizado para selecionar as condições experimentais que permitam a otimização dos procedimentos de estimação de parâmetros[83-85]. Os resultados registrados na literatura com a forma linear da Equação 10 indicam que os valores da razão de reatividade dependem da forma com que é escolhido o monômero 1. Além disso, este procedimento conduz algumas vezes a valores negativos da razão de reatividade, o que é fisicamente impossível. Existem trabalhos dedicados exclusivamente à determinação da razão de reatividade em sistemas de copolimerização. Greenley[86] recalculou a razão de reatividade de inúmeros pares monoméricos usando o método de Kelen-Tüdõs. Mark e Norman[87] catalogaram os parâmetros da razão de reatividade para um grande número de copolímeros formados por acetato de vinila (M1) e comonômeros (M2).
Modelo penúltimo
Quando o modelo penúltimo é usado para descrever a cinética de propagação, a composição do copolímero pode ser descrita como,

onde as pseudo-razões de reatividade são definidas como:

onde,

O modelo penúltimo é utilizado em sistemas em que o modelo terminal não é capaz de descrever satisfatoriamente a velocidade de polimerização. Coote e Davis[73] apresentaram uma extensa lista de pares monoméricos em que o modelo terminal mostrou-se falho. Embora o efeito penúltimo seja importante para descrever tanto a composição quanto a cinética de polimerização de alguns sistemas poliméricos, poucos parâmetros cinéticos para o modelo penúltimo estão registrados na literatura. Por apresentarem forte efeito penúltimo, as polimerizações de estireno/acrilonitrila e estireno/metacrilato de metila são exemplos típicos da aplicação bem sucedida do modelo penúltimo, tendo sido estudados por Burke et al.[88].
Monitoramento de Reações de Polimerização
O desenvolvimento de sensores para medição em linha durante uma polimerização requer um esforço multidisciplinar: modelagem matemática e processamento de dados, entendimento e conhecimento do processo, projeto do reator, técnicas de controle avançado e instrumentação. Muitos problemas encontrados em controle de reatores de polimerização podem ser atribuídos à falta de instrumentação analítica robusta e de sensores que façam medidas em linha no decorrer da polimerização. Por exemplo, as técnicas disponíveis de monitoramento da conversão são freqüentemente complicadas pela heterogeneidade ou pela natureza viscosa dos sistemas de polimerização e pela não-linearidade associada à presença de mais de uma fase, incluindo partículas de polímero de diferentes tamanhos.
Com o uso de computadores de processo na maioria dos processos industriais, enormes quantidades de dados podem ser coletadas e armazenadas para inúmeras variáveis. Temperaturas, vazões, pressão e medidas de concentração são usualmente medidas em pequenos tempos de amostragem. Porém, importantes variáveis relacionadas à qualidade e à produtividade, como massa molar e composição do polímero e rendimento do processo, normalmente só podem ser medidas fora de linha, estando muitas vezes disponível apenas após horas ou até mesmo dias depois da amostragem[89]. Nesse contexto, o monitoramento e o diagnóstico da operação dos processos foram as principais causas que levaram ao desenvolvimento teórico e industrial de "sistemas inteligentes", que consistem na junção de técnicas computacionais, métodos matemáticos e estatísticos capazes de predizer de forma satisfatória o desempenho do processo[90,91].
Nos últimos anos o desenvolvimento de sensores para os processos da Engenharia Química tornou-se bastante intenso. Dispositivos acoplados a microcomputadores revelaram-se como ferramentas bastante úteis para a medição em linha de propriedades poliméricas e, conseqüentemente, como instrumentos auxiliares no controle de reações poliméricas. Recentes desenvolvimentos de técnicas de medição em linha, especialmente as que combinam fibras óticas, oferecem enormes vantagens como: coleta de dados de inúmeras variáveis em pequenos tempos de amostragem; possibilidade de fazer medidas de reflexão sobre amostras sólidas sem qualquer pré-tratamento, manipulação mínima de amostras e habilidade de obter informações de amostra complexas; medidas in situ para um grande número de aplicações, mesmo em ambientes remotos e hostis (alta temperatura, tóxicos, explosivos, alta pressão ou de difícil acesso). Técnicas de fluorescência, de reflexão ultravioleta (UV), de espectroscopia de Raman, de espectroscopia do infravermelho próximo (NIRS), quando implementadas via sonda de fibras óticas, são ferramentas poderosas para o monitoramento in situ de reações de polimerização.
Os processos de polimerização são caracterizados por apresentarem fortes não-linearidades, significativa demora no tempo das medidas das propriedades a serem controladas e existência de perturbações não medidas, dada a enorme sensibilidade de certas reações à presença de impurezas. A falta de sensores para medida instantânea das propriedades a serem controladas (por exemplo, massas molares) torna o controle quase impossível[52,92].
Vários sensores e técnicas foram desenvolvidos para monitoramento das propriedades dos polímeros e estão disponíveis na literatura. Destacam-se as medidas de viscosidade, tensão superficial, densidade, índice de refração, composição, concentração, calorimetria, espalhamento de luz, constante dielétrica e distribuição de massa molar. Devem também ser considerados os métodos cromatográficos, os métodos espectroscópicos, os métodos baseados em estimação de estados, os métodos baseados em filtros e os métodos ultra-sônicos. Alguns trabalhos apresentam uma abordagem mais detalhada dos métodos e dos sensores para medição utilizados[93-95].
Em polimerizações tradicionais, o controle é conduzido através de amostragens, caracterização fora de linha da qualidade do polímero em um laboratório e registro manual de resultados. Esta prática persiste ainda hoje, mesmo quando algumas propriedades são difíceis de medir até mesmo fora de linha. Recentemente têm sido feitos avanços no desenvolvimento de equipamentos capazes de fazer medições em linha. Contudo, o uso destes novos sensores fez-se na maioria das vezes apenas para o monitoramento em laboratório e/ou reatores de plantas piloto. Novos desenvolvimentos em dispositivos aliados a fibras óticas, particularmente baseados em espectroscopia do infravermelho próximo (NIRS), têm aumentado bastante o horizonte para o uso de novas técnicas de monitoramento da qualidade do polímero[1].
Em sistemas onde existe a presença de mais de uma espécie reativa, a medição das propriedades pode depender da composição do copolímero. A relação entre a quantidade monitorada e a conversão do monômero normalmente requer o uso de um modelo seguro para predição da composição do polímero como uma função da conversão ou, então, a implementação de sensores em linha adicionais para determinação da conversão de cada espécie e/ou da composição do copolímero. Por outro lado, as medições em linha de propriedades moleculares, como a composição do copolímero, a distribuição de massas molares, o grau de ramificação, etc., são usualmente fracamente ligadas a variáveis operacionais do reator de polimerização. Infelizmente uma instrumentação analítica robusta e sensores para prover medições em linha de propriedades moleculares geralmente ou não são disponíveis ou requerem tempo excessivo para efetivar as medições. Para piorar, algumas vezes essas medidas são pouco confiáveis.
Os recentes desenvolvimentos das sondas de fibra ótica são apropriados para coleta remota de dados tem aumentado a possibilidade de fazer medidas in situ para um grande número de aplicações, mesmo em ambientes remotos e hostis. Isso inclui alta temperatura, ambientes tóxicos, explosivos, alta pressão ou pontos de difícil acesso. Sensores de fibra ótica também oferecem vantagens com relação às formas de configuração. Por exemplo, várias sondas podem ser montadas em diferentes correntes do processo e/ou podem ser configuradas para enviar sinais para um espectrofotômetro ou um sistema de manipulação de dados[95].
Monitoramento através de espectroscopia de infravermelho próximo (NIRS)
A espectroscopia de infravermelho próximo (NIRS) vem sendo amplamente utilizada em vários campos da indústria química para propósito de controle analítico. Isto é principalmente o resultado de vantagens substanciais desta técnica em relação a outras técnicas analíticas alternativas, destacando-se a simplificação e velocidade de obtenção de registro de espectros, a manipulação mínima de amostras e a habilidade de obter informações de amostras complexas, além da possibilidade de fazer medidas de reflexão sobre amostras sólidas sem qualquer pré-tratamento[96]. Também, esta técnica permite que grandes quantidades de informações sejam obtidas com facilidade.
As técnicas de monitoramento baseadas em NIRS são muito bem adaptadas para prover dados com estruturas simples de hardware e de software. Uma sonda de fibra ótica pode ser inserida no sistema reacional sem modificações substanciais de hardware. Geralmente, o espectro NIRS é o resultado da absorção de luz pelas moléculas orgânicas. As bandas de absorção são, na maior parte, resultados de reflexo colorido ou combinações de bandas de absorbância fundamental da região do infravermelho[26].
NIRS tem sido empregada em polimerizações para determinação in-situ de dados cinéticos e de propriedades macromoleculares de polímeros. Várias propriedades de interesse, como, por exemplo, a massa molar, a composição química, a conversão monomérica e a concentração de monômeros residuais podem ser determinadas com o auxílio da técnica NIRS. Aplicações adicionais incluem o monitoramento e controle em tempo real do tamanho de partículas de polímero[24, 26] em suspensão e da formação de partículas com estrutura do tipo casca-núcleo em processos de polimerização do tipo suspensão/emulsão[97]. A aplicação da técnica NIRS em processos de polimerização em suspensão para fins de monitoramento e/ou controle de tamanho de partículas e outras propriedades morfológicas de interesse torna-se viável graças aos fenômenos de reflexão, refração e difração na superfície das partículas. Neste caso, as elevadas taxas de espalhamento de luz (usualmente considerado como uma desvantagem para aplicação desta técnica), quando comparadas às taxas de absorção, exercem um papel fundamental para determinação de características morfológicas em sistemas de polimerização heterogêneos[96]. No que tange às aplicações de técnicas de NIRS para fins de monitoramento e controle em sistemas de polimerização em suspensão, é preciso destacar os estudos pioneiros desenvolvidos por Santos et al.[24-26]. Destacam-se também os resultados inéditos obtidos por Lenzi et al.[97], cuja aplicação de NIRS permitiu o monitoramento em linha de características morfológicas das partículas projetadas especialmente para uso como suporte para enzimas em aplicações biotecnológicas. Maiores detalhes sobre a aplicação de NIRS em diversos sistemas de polimerização (massa, solução, suspensão, emulsão) podem ser obtidos no trabalho publicado recentemente por Santos et al.[96].
Técnicas de calibração multivariável para o NIRS admitem uma relação linear entre os parâmetros e a intensidade de absorção das bandas de espectros resultantes de derivações de primeira e segunda ordem[98]. Segundo Blanco et al.[99], as redes neurais artificiais podem ser usadas para superar as não-linearidades. Contudo, Santos[24] mostrou que o uso de redes neuronais em nada melhora o desempenho de modelos de calibração de NIRS desenvolvidos para polimerizações de estireno em suspensão. Santos et al.[24, 26] monitorou o tamanho médio das partículas de estireno em um processo de polimerização em suspensão usando NIRS. Para isso utilizou um modelo baseado no método de projeção PLS (Partial Least Squares Mínimos Quadrados Parciais). Umas das principais características da PLS é que pode ser usada para montar modelos que relacionem mais de uma variável predita para muitas variáveis de estado[89,100-102].
De acordo com Machado[103] a técnica NIRS pode ser empregada para controle de composição em copolimerizações em suspensão de ácido acrílico/acetato de vinila. As Figuras 3 e 4 ilustram a predição da concentração de ácido acrílico e acetato de vinila através do monitoramento em linha via NIRS durante a copolimerização. Observa-se que as concentrações dos monômeros podem ser monitoradas em linha; conseqüentemente, técnicas de controle de processo podem ser utilizadas conjugadas com a técnica NIRS para fins de controle da composição do copolímero em tempo real em processos de polimerização em suspensão.
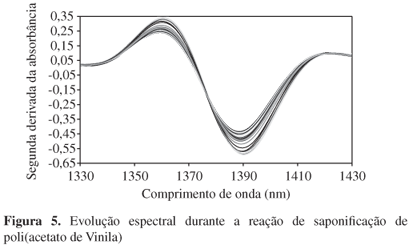
A técnica NIRS também pode ser empregada com sucesso para acompanhamento do grau de hidrólise de partículas de poli(álcool vinílico) PVA obtidas da saponificação de poli(acetato de vinila) PVAc em processo em suspensão. Recentemente, Peixoto et al.[3] desenvolveram um novo processo de obtenção de poli(álcool vinílico) em suspensão, capaz de formar PVA na forma de partículas esféricas de PVA/PVAc, apresentando estrutura casca-núcleo, visando a aplicação desse material em processo de embobilização vascular[3]. O grau de saponificação final do material polimérico pode ser controlado durante a polimerização através da manipulação de variáveis de processo, como por exemplo, a concentração de NaOH no meio reacional, o tempo de saponificação e o tamanho das partículas precursoras de PVAc. A Figura 5 mostra os espectros de NIR tomados durante uma reação de saponificação, indicando que o teor de grupos hidroxilas podem ser monitorados durante o processo de saponificação, favorecendo o uso da técnica NIRS para o controle do grau de hidrólise do material polimérico final.
Conclusão
Aspectos importantes dos processos de polimerização em suspensão foram revistos. Foi realizada uma descrição dos principais processos de polimerização em suspensão empregados para obtenção de diferentes materiais poliméricos. O efeito das principais variáveis de processo, como por exemplo, agentes de suspensão, iniciadores, eletrólitos, natureza do monômero, etc, e de fenômenos típicos de processos de polimerização em suspensão, como os efeitos gel e vítreo, a quebra e a coalescência das partículas poliméricas são discutidos extensamente. Além disso, são mostrados alguns exemplos da utilização da técnica de espectroscopia de infravermelho próximo para fins de monitoramento e controle de processos de polimerização, visando a determinação em tempo real da concentração de espécies monoméricas no meio, do tamanho médio das partículas poliméricas e de características morfológicas do material polimérico.
Enviado: 02/02/07
Reenviado: 20/03/07
Aceito: 23/03/07
- 1.Kiparissides, C. - Chem. Eng. Sci., 51, p.1637 (1996).
- 2.Dowding, P. J. & Vicent, B. - Coll. Surf. A: Physicochem. Eng. Asp., 161, p.259 (2000).
- 3.Peixoto, L. S.; Machado, F.; Niemeyer, M. A. L.; Espinosa, G.; Melo, P. A.; Nele, M. & Pinto, J. C. - Macromol. Symp., 243, p.190 (2006).
- 4.Lenzi, M. K.; Machado, F.; Lima, E. L.; Pinto, J. C. - J. Appl. Polym. Sci., 89, p.3021 (2003).
- 5.Santos, J. G. F.; Peixoto, L. S.; Nele, M.; Melo, P. A. & Pinto, J. C. - Macromol. Symp., 243, p.1 (2006).
- 6.Lemos, L.; Nele, M.; Melo, P. & Pinto, J. C. - Macromol. Symp., 243, p.13-23 (2006).
- 7.Yuan, H. G.; Kalfas, G. & Ray, W. R. - Macromol. Sci. - Rev. Macromol. Chem. Phys., C31, p.215 (1991).
- 8.Tang, L. G.; Weng, Z. X. & Pan, Z. R. - Eur. Polym. J., 32, p.1139 (1996).
- 9.Askari, F.; Nafisi, S.; Omidian, H. & Hashemi, S. A. - J. Appl. Polym. Sci., 50, p.1851 (1993).
- 10.Omidian, H.; Hashemi, S. A. & Nafisi, S. - J. Appl. Polym. Sci., 54, p.241 (1994).
- 11.Liu, Z. & Brooks, B. W. - Polymer, 40, p.2181 (1999).
- 12.Lenzi, M. K.; Lima, E. L. & Pinto, J. C. - Polim. Cienc. Tecnol., 14, p.112 (2004).
- 13.Lenzi, M. K.; Cunningham, M. F.; Lima, E. L. & Pinto, J. C. - J. Appl. Polym. Sci., 96, p.1950 (2005).
- 14.Gu, Q.; Lin, Q.; Hu, C. & Yang, B. - J. Appl. Polym. Sci., 95, p.404 (2005).
- 15.Odian, G. - "Principles of Polymerization", New York: Wiley-Interscience (2004).
- 16.Araújo, P. H. H. & Pinto, J. C. - Braz. J. Chem. Eng., 17, p.383 (2000).
- 17.Sáenz, J. M. & Asua, J. M. - Coll. Surf. A: Physicochem. Eng. Asp., 153, p.61 (1999).
- 18.Cao, K.; Yu, J.; Li, B. G.; Li, B. F. & Pan, Z. R. - Chem. Eng. J., 78, p.211 (2000).
- 19.Cunningham, M. F. - Polym. React. Eng., 7, p.231 (1999).
- 20.Schork, F. J.; Luo, Y.; Smulders, W.; Russum, J. P.; Butté, A. & Fontenot, K. - Adv. Polym. Sci., 175, p. 129 (2005).
- 21.Pollaco, G.; Semino, D. & Palla, M. - Polym. Eng. Sci., 36, p.2088 (1996).
- 22.Chatzi, E. G. & Kiparissides, C. - Chem. Eng. Sci., 49, p.5039 (1994).
- 23.Lazrak, N.; Bolay, N. L. E. & Ricard, A. - Eur. Polym. J. 34, p.1637 (1998).
- 24.Santos, A. F.; Lima, E. L. & Pinto, J. C. - J. Appl. Polym. Sci., 70, p.1737 (1998).
- 25.Santos, A. F.; Lima, E. L. & Pinto, J. C. - Polim. Cienc. Tecnol., p.39 (1999).
- 26.Santos, A. F.; Lima, E. L. & Pinto, J. C. - J. Appl. Polym. Sci., 77, p.453 (2000).
- 27.Kalfas, G.; Yuan, H. & Ray, W. H. - Ind. Eng. Chem. Res., 32, p.1831 (1993).
- 28.Shaghaghi, S.; Mahdavian, A. R. - Polym. Plast. Technol. Eng. 45, p.109 (2006).
- 29.Georgiadou, S. & Brooks, B. - Chem. Eng. Sci., 61, p.6892 (2006).
- 30.Jonsson, M.; Nordin, O.; Malmstrom, E. & Hammer, C. - Polymer, 47, p.3315 (2006).
- 31.Nunes, D. S. S. & Coutinho, F. M. B. - Eur. Polym. J., 38, p.1159 (2002).
- 32.Araújo, O. - Estudo Experimental e Modelagem Matemática do Processo de Polimerização em Emulsão de Acetato de Vinila, 1992, Escola Politécnica da USP, SP, Brasil.: São Paulo.
- 33.Mark, F. G.; Bikales, N. M.; Overberger, C. G. & Menges, G. - "Encyclopedia of Polymer Science Technology", New York: John Wiley & Sons (1989).
- 34.Lee, S. G.; Kim, J. P.; Kwon, I. C.; Shin, D. S.; Han, S. S. & Lyoo, W. S. - J. Appl. Polym. Sci., 101, p.4064 (2006).
- 35.Wolff, E. H. P.; René Bos, A. N. - Ind. Eng. Chem. Res., 36, p.1163 (1997).
- 36.Dotson, N. A.; Galván, R.; Laurence, R. L. & Tirrel, M. - "Polymerization Process Modeling", New York: VCH Publishers (1996).
- 37.Kresser, T. O. J. - "Polyolefin Plastics", New York: Van Nostrand Reinhold Company (1969).
- 38.Dubé, M. A.; Soares, J. B. P.; Pendilis, A.; Hamielec, A. E. - Ind. Eng. Chem. Res., 36, p.966 (1997).
- 39.Tsouris, C. & Tavlarides, L. L. - AIChE J., 40, p.395 (1994).
- 40.Wright, H. & Ramkrishna, D. - AIChE J., 40, p.767 (1994).
- 41.Jahanzad, F.; Sajjadi, S. & Brooks, B. W. - Ind. Eng. Chem. Res., 44, p.4112 (2005).
- 42.Kotoulas, C. & Kiparissides, C. - Chem. Eng. Sci., 61, p.332 (2006).
- 43.Abu-Ayana, Y. & Mohsen, R. - Polym. Plast. Technol. Eng., 44, p.1503 (2005).
- 44.Lyoo, A. S.; Park, C. S.; Choi, K. H.; Kwak, J. W.; Yoon, W. S. & Noh, S. K. - Polym. Plast. Technol. Eng., 44, p.475 (2005).
- 45.Vivaldo-Lima, E.; Wood, P. E.; Hamielec, A. E. & Penlidis, A. - Ind. Eng. Chem. Res., 36, p.939 (1997).
- 46.Mikos, A. G.; Takoudis, C. G. & Peppas, N. A. - J. Appl. Polym. Sci., 31, p.2647 (1986).
- 47.Machado, R. A. F.; Pinto, J. C.; Araújo, P. H. H. & Bolzan, A. - Braz. J. Chem. Eng., 17, p.395 (2000).
- 48.Alvarez, J.; Alvarez, J. & Hernandez, M. - Chem. Eng. Sci., 49, p.99 (1994).
- 49.Maggioris, D.; Goulas, A.; Alexopoulos, A. H.; Chatzi, E. G. & Kiparissides, C. - Comp. Chem. Eng., p.S315 (1998).
- 50.Maggioris, D.; Goulas, A.; Alexopoulos, A. H.; Chatzi, E. G. & Kiparissides, C. - Chem. Eng. Sci., 55, p.4611 (2000).
- 51.Kalfas, G.; Yuan, H.; Ray, W. H. - Ind. Eng. Chem. Res., 32, p.1822 (1993).
- 52.Crowley, T. J. & Choi, K. - J. Proc. Cont., 6, p.119 (1996).
- 53.Chiu, W. Y.; Carrat, G. M. & Soong, D. S. - Macromolecules, 16, p.348 (1983).
- 54.Maschio, G.; Bello, T. & Scali, C. - Chem. Eng. Sci., 49, p.5071 (1994).
- 55.Sharma, D. K. & Soane, D. S. - Macromolecules, 21, p.700 (1988).
- 56.Ray, A. B.; Saraf, D. N. & Gupta, S. K. - Polym. Eng. Sci., 35, p.1290 (1995).
- 57.Dua, V.; Saraf, D. N. & Gupta, S. K. - J. Appl. Polym. Sci., 59, p.749 (1996).
- 58.Srinivas, T.; Sivakumar, S.; Gupta, S. K. & Saraf, D. N. - Polym. Eng. Sci., 36, p.311 (1996).
- 59.Nagasubramanian, K. & Graessley, W. W. - Chem. Eng. Sci., 25, p.1549 (1970).
- 60.Baade, W.; Moritz, H. U. & Reichert, K. H. - J. Appl. Polym. Sci., 27, p.2249 (1982).
- 61.Friis, N. & Nyhagen, L. - J. Appl. Polym. Sci., 17, p.2311 (1973).
- 62.Yoo, K. Y.; Jeong, B. G. & Rhee, H. K. - Ind. Eng. Chem. Res., 38, p.4805 (1999).
- 63.Pinto, J. C. & Ray, W. H. - Chem. Eng. Sci., 36, p.367 (1995).
- 64.Schmidt, A. D. & Ray, W. H. - Chem. Eng. Sci., 36, p.1401 (1981).
- 65.Oneil, G. A.; Wisnudel, M. B. & Torkelson, J. M. - Macromolecules, 31, p.4537 (1998).
- 66.Oneil, G. A.; Wisnudel, M. B. & Torkelson, J. M. - AIChE J., 44, p.1226 (1998).
- 67.Zhang, S. X. & Ray, W. H. - Ind. Eng. Chem. Res., 36, p.1310 (1997).
- 68.Machado, F.; Lima, E. L. & Pinto, J. C. - J. Appl. Polym. Sci., 93, p.1077 (2004).
- 69.Machado, F.; Lima, E. L. & Pinto, J. C. - Ind. Eng. Chem. Res., 43, p.7324 (2004).
- 70.Machado, F.; Lima, E. L. & Pinto, J. C. - Ind. Eng. Chem. Res., 43, p.7312 (2004).
- 71.Crevecoeur, J. J.; Nelissen, L. & Lemstra, P. J. - Polymer, 40, p.3685 (1999).
- 72.Kawaguchi, Y.; Itamura, Y.; Onimura, K. & Oishi, T. - J. Appl. Polym. Sci., 96, p.1306 (2005).
- 73.Coote, M. L. & Davis, T. P. - Prog. Polym. Sci., 24, p.1217 (1999).
- 74.Johnson, A. F. - "Uses and Applications of Polymers". Polymer Reactor Engineering, ed. McGreavy, C., New York: VCH Publishers1 (1994).
- 75.Lu, Q.; Weng, Z. X.; Shan, G. R.; Lai, G. Q. & Pan, Z. R. - J. Appl. Polym. Sci., 101, p.4270 (2006).
- 76.Cavalcanti, M. J. R. & Pinto, J. C. - J. Appl. Polym. Sci., p.1683 (1997).
- 77.Lu, Q.; Shan, G. R.; Weng, Z. X. & Huang, Z. M. - Acta Polym. Sin., 2, p.258 (2006).
- 78.Lu, Q.; Weng, Z.; Zhou, S.; Huang, Z. & Pan, Z. - Eur. Polym. J., 38, p.1337 (2002).
- 79.Billmeyer, F. W. - "Textbook of Polymer Science", New York: John Wiley & Sons (1984).
- 80.Rosen, S. L. - "Fundamental Principles of Polymeric Materials", New York: John Wiley & Sons (1993).
- 81.Finemam, M. & Ross, S. D. - J. Polym. Sci., 5, p.259 (1950).
- 82.Kelen, T. & Tüdõs, F. - J. Macromol. Sci. - Chem., A9, p.1 (1975).
- 83.Behnken, D. W. - J. Polym. Sci.: Part A, 2, p.645 (1964).
- 84.Tidwell, P. W. & Mortimer, G. A. - J. Polym. Sci.: Part A, 3, p.369 (1965).
- 85.Tidwell, P. W. & Mortimer, G. A. - J. Macromol. Sci. - Rev. Macromol. Chem., C4, p.281 (1970).
- 86.Greenley, R. Z. - J. Macromol. Sci. - Chem., A14, p.445 (1980).
- 87.Mark, F. G. & Norman, N. G. - "Encyclopedia of Polymer Science Technology", New York: John Wiley & Sons (1971).
- 88.Burke, A. L.; Duever, T. A. & Pendilis, A. - Ind. Eng. Chem. Res., 36, p.1016 (1997).
- 89.Macgregor, J. F.; Jaeckle, C.; Kiparissides, C. & Koutoud, M. - AIChE J., 40, p.826 (1994).
- 90.Stephanopoulos, G. & Han, C. - Comp. Chem. Eng., 20, p.743 (1996).
- 91.Çetinkaya, S.; Zeybek, Z.; Hapoglu, H. & Alpbaz, M. - Comp. Chem. Eng., 30, p.1315 (2006).
- 92.Gattu, G. & Zafiriou, E. - Chem. Eng. J., 75, p.21 (1999).
- 93.Chen, D. C. H. & Pendilis, A. - J. Macromol. Sci. - Rev. Macromol. Chem. Phys., C30, p.1 (1990).
- 94.Macgregor, J. F.; Pendilis, A. & Hamielec, A. E. - Polym. Proc. Eng., 2, p.179 (1984).
- 95.Kammona, O.; Chatzi, G. E. & Kiparissides, C. - J. Macromol. Sci. - Rev. Macromol. Chem. Phys., C39, p.57 (1999).
- 96.Santos, A. F.; Machado, F.; Lenzi, M. K. & Pinto, J. C. - Polym. Plast. Technol. Eng., 44, p.1 (2005).
- 97.Lenzi, M. K.; Lima, E. L. & Pinto, J. C. - J. Near Infrared. Spectrosc., 14, p.179 (2006).
- 98.Bertran, E.; Blanco, M.; Maspoch, S.; Ortiz, M. C.; Sánchez, M. S. & Sarabia, L. A. - Chem. Intell. Lab. Syst., 49, p.215 (1999).
- 99.Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S. & Pagés, J. - Anal. Chim. Acta, 384, p.207 (1999).
- 100.Wise, B. M. & Gallagher, N. B. - J. Proc. Cont., 6, p.329 (1996).
- 101.Kourti, T.; Lee, J. & Macgregor, J. F. - Comp. Chem. Eng., 20, p.S745 (1996).
- 102.Sánchez, M. S.; Bertran, E.; Sarabia, L. A.; Ortiz, M. C.; Blanco, M. & Coel ho, J. - Chem. Intell. Lab. Syst., 53, p.69 (2000).
- 103.Machado, F., "Modelagem e Controle da Composição em Sistema de Polimerização em Suspensão" 2002, Tese de M.Sc, COPPE/UFRJ: Rio de Janeiro, Brasil.
Datas de Publicação
-
Publicação nesta coleção
17 Set 2007 -
Data do Fascículo
Jun 2007
Histórico
-
Aceito
23 Mar 2007 -
Recebido
02 Fev 2007