Abstract
Chitosan/gelatin microparticles were prepared by complex coacervation. Three chitosan (CH) samples, with different acetylation degrees and intrinsic viscosities, were used together with commercial gelatin (G) samples. Microparticles formation was investigated at various CH/G ratios, within the pH range of 3.5 to 6.0. Reactions were carried out at 40 and 60 ºC, for 2, 4, and 6 hours. Turbidity measurements performed at 633 nm were used to monitor the process. The resulting curves revealed maximum values, which were correlated to the glucosamine content of CH samples. After isolation, yields were determined, and the microparticles were characterized by infrared spectroscopy (FTIR) and thermogravimetry (TGA). Both techniques evidenced the formation of coacervate microparticles. The highest yields in microparticles were determined for the system comprising the CH sample with the lowest degree of acetylation and intrinsic viscosity, and the gelatin sample with the lowest bloom strength.
Chitosan; gelatin; microparticles; coacervation
ARTIGO TÉCNICO CIENTÍFICO
Evaluating conditions for the formation of chitosan/gelatin microparticles
Marcia C. Silva; Cristina T. Andrade
Programa Ciência de Alimentos, Instituto de Química, UFRJ
Autor para correspondênciaAutor para correspondência: Cristina T. Andrade Instituto de Macromoléculas Professora Eloísa Mano, UFRJ, Centro de Tecnologia, Bloco J Caixa Postal 68525 CEP: 21945-970, Rio de Janeiro, RJ, Brasil E-mail: ctandrade@ima.ufrj.br
ABSTRACT
Chitosan/gelatin microparticles were prepared by complex coacervation. Three chitosan (CH) samples, with different acetylation degrees and intrinsic viscosities, were used together with commercial gelatin (G) samples. Microparticles formation was investigated at various CH/G ratios, within the pH range of 3.5 to 6.0. Reactions were carried out at 40 and 60 ºC, for 2, 4, and 6 hours. Turbidity measurements performed at 633 nm were used to monitor the process. The resulting curves revealed maximum values, which were correlated to the glucosamine content of CH samples. After isolation, yields were determined, and the microparticles were characterized by infrared spectroscopy (FTIR) and thermogravimetry (TGA). Both techniques evidenced the formation of coacervate microparticles. The highest yields in microparticles were determined for the system comprising the CH sample with the lowest degree of acetylation and intrinsic viscosity, and the gelatin sample with the lowest bloom strength.
Keywords:Chitosan, gelatin, microparticles, coacervation.
Introduction
Complex coacervation is a liquid-liquid phase separation process, which occurs spontaneously from mixed solutions of oppositely charged polyelectrolytes. The system consists of two immiscible phases; an insoluble polymer-rich phase that contains the polyelectrolytes complex stabilized mainly by electrostatic interaction, and a solvent-rich phase. Phase separation of insoluble coacervates was shown to be preceded by the formation and aggregation of nearly neutral soluble complexes[1-3].
Experimentally, the coacervation process is conducted under mild conditions, and the microencapsulation of labile or easily damaged substances can be achieved. The interest on such process is based on its technological applications in several industrial sectors. In the medical, pharmaceutical, and food sectors, biocompatibility, biodegradability and nontoxicity are required, which reduces the number of potentially available polyelectrolytes to be used. Because of these requirements, polysaccharides and proteins have been employed extensively to encapsulate biologically active substances by complex coacervation. For example, results from systems such as gelatin-gum acacia[4,5], gelatin-heparin[6,7], gelatin-carboxymethyl cellulose[8], β-lactoglobulin-gum acacia[9], β-lactoglobulin-pectin[10], vegetal protein-gum acacia[11] have been reported. In general, the protein is used below its isoelectric point with an anionic polysaccharide, at pHs not low enough to suppress the dissociation of the anionic polysaccharide.
In the present study, experimental samples of chitosan, and commercial samples of type-B gelatin were mixed under different conditions, to investigate the formation of coacervate microparticles. Chitosans consist of a family of polysaccharides obtained by partial deacetylation of chitin, and constituted by (1,4)-β-linked 2-acetamide-2-deoxy-D-glucose, and mainly of its N-deacetylated repeating units. Upon dissolution in acid pH, the amino groups of chitosan become protonated, and the polymer behaves as a polycation[12]. Gelatin is a polyampholyte obtained from denatured collagen. Depending on the hydrolysis process, gelatin has different physical properties. The primary objective of this study was to investigate yields in chitosan/gelatin coacervate microparticles, aiming their application as wall materials in microencapsulation. To the authors' knowledge, only a few papers were published on complex coacervation of gelatin/chitosan systems; type-B gelatin-chitosan glutamate[13] and type-A gelatin-chitosan systems[14]. In the present case, type-B gelatin samples were used.
Materials and Methods
Chitosan samples were obtained by partial deacetylation of chitin from shells of Penaeus schmitti shrimp. For demineralization, shells were treated with 0.25 M HCl for 30 minutes. Chitin deproteinization was performed under stirring with 1 M NaOH at room temperature, for 24 hours. Pigment elimination was achieved by immersing the sample in acetone and ethyl alcohol baths.
Deacetylation of chitin was performed in two stages. In the first stage, chitin powder was reacted under stirring with 10% (w/v) NaOH solution at room temperature, for 5 hours. The reaction product was neutralized by successive washings in baths of deionized water, filtered, and dried at 50 ºC for 12 hours. The second stage was carried out at 100 ºC under nitrogen bubbling, in the presence of sodium borohydride (0.1 g/g of deacetylated chitin), under different reaction conditions (Table 1). Three chitosan samples (CH1, CH2, and CH3) were obtained, and characterized by 1H NMR and by viscosity measurements, which provided data, respectively, of acetylation degrees (DA), and intrinsic viscosity ([η]), also shown in Table 1. Both DA and [η] may be considered as chitosan most important properties.
Acetylation degrees (DA) were determined by 1H NMR spectrometry. Spectra were acquired in a Varian Mercury VX 300 (300 MHz for 1H) (Palo Alto, USA), equipped with a 5 mm Indirect Detection probe. Chitosan samples (15 mg) were dissolved in a mixture of 1.96 mL D2O and 0.023 mL of 35% DCl (Cambridge Isotope Laboratories, Andover, USA) by stirring at room temperature for 3 hours. Spectra were recorded at 70 ºC with 90º pulse that corresponded to a pulse width of 11 µs, 6 seconds of delay time, and an acquisition time of 2 seconds. At least, 100 scans were acquired. DA values were determined from the following Equation 1.

where ICH3 is the intensity of the methyl protons signal of the β-(1,4)-2-deoxy-2-acetamido-D-glucose residues, and IH2-H6' is the sum of the intensities of H-2 of the deacetylated residue and H-2 to H-6' signals from both residues.
Viscosity measurements were performed with a Ubbelohde capillar viscosimeter (inner diameter of 0.58 mm) at 25 ± 1 ºC. Solutions were prepared with 0.3 M acetic acid/0.2 M sodium acetate (pH 4.5) by stirring at room temperature for 12 hours. Before measuring, the solutions were centrifuged at 2000 rpm for 20 minutes. Intrinsic viscosities were determined by extrapolation to zero concentration according to the Huggins equation.
Type-B gelatin samples, denoted G1, G2, and G3, respectively, of bloom strength 90-100 (Mw 20,000-25,000), bloom strength 175 (Mw 40,000-50,000), and bloom strength 300 (Mw 50,000-100,000) were purchased from Sigma-Aldrich Brasil Ltda. (São Paulo, SP).
Chitosan (CH) solutions at 2 g.L-1 were prepared in 1% (v/v) acetic acid under stirring at ambient temperature for 12 hours. Gelatin (G) solutions at 2, 10 and 20 g.L-1 concentrations were prepared in Milli-Q water under stirring at 40 ºC for 30 minutes. To prepare coacervate microparticles, 10 mL of CH solution were added dropwise to 10 mL G solution under stirring. The systems CH1/G3, CH2/G2, and CH3/G1 were investigated. Depending on the concentration of G solution, the CH/G ratios were 1:1, 1:5, or 1:10. The pH of the mixtures was adjusted with 0.1 M HCl or 0.1 M NaOH. The reaction conditions were maintained for 2, 4, or 6 hours, at 40 and 60 ºC.
Reactions were followed by spectrophometric measurements in a UV-Vis Varian spectrophotometer model Cary100 (Palo Alto, CA, USA), using a matched pair of quartz cuvettes with a 1 cm path length. The spectral measurements were taken in the percent transmittance mode (% T), at 633 nm[15]. The turbidity index (100 - %T at 633 nm) was plotted against pH.
Proportionally higher quantities of reagents were used to prepare microparticles for yield determinations and characterization by infrared spectroscopy (FTIR) and thermogravimetric analysis (TGA). After centrifuging (2000 rpm for 10 minutes), the clear supernatant was decanted, the sediment was washed with water, and redispersed. This procedure was repeated three times, the coacervates were dried to constant weight in a desiccator, and weighed (m3). Yields were calculated in relation to the corresponding sum of chitosan (m1) and gelatin (m2) masses used in each experiment.
FTIR spectra were obtained in a Perkin-Elmer 1720 X spectrometer (Beaconsfield, UK), in KBr disks, with 20 scans, at a resolution of 2 cm-1, for CH, G, and dry CH/G coacervate microparticles. Coacervate samples were analyzed in a TGA Q500 equipment from TA Instruments (New Castle, DE, USA) under nitrogen atmosphere. Samples (12 mg) were heated from 25 to 700 ºC at a heating rate of 20 ºC/min.
Results and Discussion
Complex coacervation is influenced by the ratio between oppositely charged polyelectrolytes, reaction temperature, reaction time, and pH. To investigate the formation of coacervates, reactions were carried out for the systems CH1/G3, CH2/G2, and CH3/G1, at CH/G ratios of 1:1, 1:5, and 1:10, at 40 and 60 ºC, for 2, 4, and 6 hours.
By using a variety of experimental techniques, several structural and morphological transitions were recognized upon the gradual change of pH[16]. The two most important of those transitions occur at different pH values, depending on the system. The critical pH (pHc) can be considered as a phase transition on the molecular scale, and indicates the formation of soluble primary macromolecular complexes. The pH of coacervate formation, pHΦ, would correspond to phase separation and observation of coacervate droplets[3].
In the present work, structural changes were observed within the pH range of 3.5 to 6.0 by spectrophotometry. Figure 1 shows the variation of turbidity as a function of pH for the three systems. At pH 6, turbidity was low for all systems. As pH was decreased, different behaviors were observed. For the CH1/G3 system, in which the chitosan sample had the highest DA, and consequently the lowest content in -NH2 groups, a slight increase in turbidity was visualized up to pH around 4.2. The slight increase in turbidity is clearly observed, mainly when the CH1/G3 ratios were 1:1 and 1:5, as in Figure 1; a1, a2, respectively. In this case, pH 4.2 may be related to pHc.
For the other systems, this molecular scale phase transition was displaced to higher pH values, as revealed for CH1/G3 at a 1:10 ratio (Figure 1a3), and CH2/G2 at all polymer ratios (Figure 1; b1, b2, b3). The increase in gelatin concentration, as in the CH1/G3 system, and in the amount of -NH2 groups, as in the CH2/G2 system, might have contributed to the appearance of the critical point at a higher pH. This behavior can be explained qualitatively from the fact that, when there is a tendency for a higher content of positively charged segments, a larger amount of negative charge is necessary to achieve charge neutralization[14,17].
For the CH3/G1 system (Figure 1; c1, c2, c3), in which the chitosan sample had the lowest DA, pHc was not so clearly detected. Nevertheless, an indication of the occurrence of pHc at an even higher pH may be envisaged in Figure 1c2, for the CH3/G1 ratio of 1:5, as expected.
The spectrophotometric method used in this work allows the estimation of a maximum, at which pH (pHΦ) the maximum interaction between the biopolymers should be occurring. This maximum could be more easily detected (at pH 4) for the CH1/G3 system (Figure 1; a1, a2, a3). Differences in degree of acetylation may explain the appearance of the maximum at pH 4.0 for the CH1/G3 system, and at a higher pH (near pH 4.5) for the other systems (Figure 1; b1, b2, b3, c1, c2, c3). It is important to notice that, for CH2/G2 and CH3/G1, other transitions may be occurring around the large range of pH where the maximum is observed. Such transitions were appointed by other techniques[18].
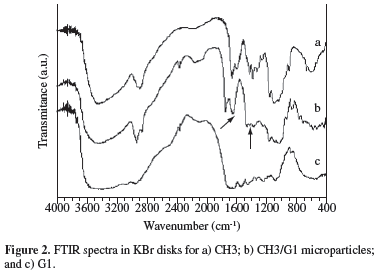
Formation of complex coacervates was investigated by FTIR. Figure 2 shows the spectra of CH3, G1 and CH3/G1 microparticles (1:5 ratio), obtained at pH 4.5, after reaction at 60 ºC for 4 hours. Qualitatively similar spectra were obtained for the other compositions. The CH3 spectrum exhibits peaks at 1652 and 1591 cm-1, attributed to C = 0 stretching (Amide I) and -NH bending (Amide II), respectively. G1 spectrum presents several bands in the 1655-1230 cm-1 range, characteristic of secondary amide groups. Evidence of CH/G complex formation comes from the appearance of the bands at 1642 cm-1 and around 1416 cm-1, attributed to -NH3+ asymmetric angular deformation of chitosan, and to symmetric stretching of COO- of gelatin, respectively[19,20].
Evidence of coacervate formation was also obtained by thermogravimetric analysis (TGA). TGA was carried out for chitosan, gelatin and coacervate microparticles samples. In Figure 3, the thermograms for CH1, G3, and for the microparticle sample obtained at 1:5 ratio, pH 4, and 60 ºC for 4 hours are shown, as an example. After loss of water, the thermal degradation of the microparticles began at an intermediate temperature (To = 285 ºC), between those of G3 (To = 272 ºC) and CH1 (To = 298 ºC). Actually, To varied for chitosan and gelatin samples, as well as for microparticles samples (Table 2). According to the results, higher contents in glucosamide groups (higher DA) favor the initiation of thermal degradation of chitosan at lower temperatures. For the microparticles samples, thermal stabilization depends on the components and their ratios. As the CH/G ratio decreases, gelatin in excess is likely to form a shell around the particles, and To decreases accordingly.
For application of coacervates as wall materials in micro-encapsulation processes, a reasonably-high yield is important. Microcapsules were isolated, exaustively washed and dried, before yields determination. Table 3 shows reaction conditions and yield results for the systems CH1/G3, CH2/G2, and CH3/G1. As expected, reactions conducted at pH ~ pHΦ, as determined by turbidimetry (Figure 1), led to higher yields in microparticles. In general, the increase in reaction time from 2 to 4 hours also contributed to higher yields. Increasing reaction temperatures from 40 to 60 ºC led to higher yields when results are compared for the same system at the same ratio between components. This result implies that mobility would favor biopolymer chains to achieve suitable conformations for complex formation. The fact that the highest yields were observed for the CH3/G1 system, formed by the chitosan and gelatin samples with the lowest molar mass, confirms that conclusion.
Conclusions
Complex coacervation between chitosan and gelatin was studied by spectrophotometry, as a function of pH. Chitosan samples with varied viscosities and degrees of acetylation were used in 1:1, 1:5, and 1:10 ratios with type-B gelatin samples differing in bloom strength. The critical pH (pHc) could be identified for some systems. Microparticles were formed and isolated by carrying out reactions with corresponding higher amounts of the polyelectrolyte and polyampholyte, at pH conditions near pHΦ (pH 4 and pH 4.5). Coacervation was evidenced by FTIR spectra, which showed the expected characteristic bands. Thermograms obtained for the resulting microparticles revealed that initiation of thermal degradation could be correlated to the chitin and gelatin samples, as well as to the ratio between them. The highest yield in microparticles was achieved by using the components with the lowest molar masses, reaction at 60 ºC, reaction time of 4 hours, and chitosan/gelatin ratio of 1:10.
Acknowledgments
The authors thank FAPERJ (Proc. E-26/151.969/2004) and CAPES-GRICES (Proc. 194/08) for financial support.
Enviado: 27/11/08
Reenviado: 02/02/09
Aceito: 09/02/09
- 1. Veis, A. & Aranyi, C. - J. Phys. Chem., 64, p.1203 (1960).
- 2. Mattison, K. W.; Brittain, I. J. & Dubin, P. L. - Biotechnol. Prog., 11, p.632 (1995).
- 3. Mekhloufi, G.; Sanchez, C.; Renard, D. & Guillemin, S. & Hardy, J. - Langmuir, 21, p.386 (2005).
- 4. Jegat, C. & Taverdet, J. L. - Polym. Bull., 44, p.345 (2000).
- 5. Junyaprasert, V. B.; Mitrevej, A.; Sinchaipanid, N.; Boonme, P. & Wurster, D. E. - Drug Developm. Ind. Pharm., 27, p.561 (2001).
- 6. Tsung, M. & D. J Burgess, D. J. - J. Pharm. Sci., 86, p.603 (1997).
- 7. Lamprecht, A.; Ubrich, N. & Maincent, P. - Eur. J. Pharm. Biopharm., 67, p.632 (2007).
- 8. Lii, C.; Tomasik, P.; Zaleska, H.; Liaw, S. & Lai, V. M.-F. - Carbohydr. Polym., 50, p.19 (2002).
- 9. Schmitt, C.; Sanchez, C.; Thomas, F. & Hardy, J. - Food Hydrocoll., 13, p.483 (1999).
- 10. Girard, M.; Turgeon, S. L. & Gauthier, S. F. - J. Agric. Food Chem., 51, p.4450 (2003).
- 11. Ducel, V.; Richard, J.; Saulnier, P.; Popineau, Y. & Boury, F. - Colloid Surf. A: Physicochem. Eng. Aspects, 232, p.239 (2004).
- 12. Torres, M. A.; Vieira, R. S.; Beppu, M. M. & Santana, C. C. - Polímeros: Ciência e Tecnologia, 15, p.306 (2005).
- 13. Remuñán-López, C. & Bodmeier, R. - Int. J. Pharm., 135, p.63 (1996).
- 14. Gupta, A. N.; Bohidar, H. B. & Aswal, V. K. - J. Phys. Chem. B, 111, p.10137 (2007).
- 15. Mitra, D.; Bhattacharya, S. C. & Moulik, S. - J. Phys. Chem. B, 112, p.6609 (2008).
- 16. Kaibara, K.; Okazaki, T.; Bohidar, H. B. & Dubin, P. L. - Biomacromolecules, 1, p.100 (2000).
- 17. Kimura, I. Y.; Gonçalves Jr., A. C.; Stolberg, J.; Laranjeira, M. C. M. & Fávere, V. T. - Polímeros: Ciência e Tecnologia, 3, p.51 (1999).
- 18. Mekhloufi, G.; Sanchez, C.; Renard, D.; Guillemin, S. & Hardy, J. - Langmuir, 21, p.386 (2005).
- 19. Günzler, H. & Böck, H. - "IR-Spektroskopie. Eine Einführung", Verlag Chemie,. Weinheim (1975).
- 20. Josué, A.; Laranjeira, M. C. M.; Fávere, V. T.; Kimura, I. Y. & Pedrosa, R. C. - Polímeros: Ciência e Tecnologia, 10, p.116 (2000).
Autor para correspondência:
Publication Dates
-
Publication in this collection
13 Aug 2009 -
Date of issue
June 2009
History
-
Accepted
09 Feb 2009 -
Received
27 Nov 2008