
Abstract
New copolymers from hindered amines and vinyl monomers were synthesized by radical chain polymerization. To obtain polymeric HALS, acrylamide-(1ATP) and acrylate-(4ATP) monomers, derivatives from 2,2,6,6-tetramethylpiperidine and 2,2,6,6-tetramethyl-4-piperidinol were synthesized. The radical chain polymerization of 1ATP with styrene (Sty) using 1-butanethiol (BTN) resulted in a copolymer with 95 units of Sty and 15 units of 1ATP. The radical chain polymerization of 1ATP and vinyl acetate (VAc) has produced only 1ATP homopolymer. In the chain polymerization of 4ATP with Sty or VAc, the hydrogen atom bonded to the nitrogen of 4ATP is labile enough to originate another radical at this site. The steric hindrance imposed by methyl groups on this bonding site hampers its reaction with other propagating species and the formation of a copolymer or network structure will be dependent on the size of the pendent group in the vinyl monomer.
Degradation; hindered amine; photooxidation; polymeric HALS; stabilizers; synthesis
TECHNICAL SCIENTIFIC PAPERS
Synthesis and characterization of copolymers from hindered amines and vinyl monomers
Marcelo Aparecido ChinelattoI; José Augusto Marcondes AgnelliII; Sebastião Vicente CanevaroloII
IDepartment of Materials Engineering SMM, University of São Paulo USP, Brazil
IIDepartment of Materials Engineering DEMa, Federal University of São Carlos UFSCar, Brazil
Corresponding author Corresponding author: Marcelo A. Chinelatto Departamento de Engenharia de Materiais, Universidade de São Paulo USP Av. João Dagnone, 1100, Campus II CEP 13563-120, São Carlos, SP, Brasil e-mail: mchinelatto@sc.usp.br
ABSTRACT
New copolymers from hindered amines and vinyl monomers were synthesized by radical chain polymerization. To obtain polymeric HALS, acrylamide-(1ATP) and acrylate-(4ATP) monomers, derivatives from 2,2,6,6-tetramethylpiperidine and 2,2,6,6-tetramethyl-4-piperidinol were synthesized. The radical chain polymerization of 1ATP with styrene (Sty) using 1-butanethiol (BTN) resulted in a copolymer with 95 units of Sty and 15 units of 1ATP. The radical chain polymerization of 1ATP and vinyl acetate (VAc) has produced only 1ATP homopolymer. In the chain polymerization of 4ATP with Sty or VAc, the hydrogen atom bonded to the nitrogen of 4ATP is labile enough to originate another radical at this site. The steric hindrance imposed by methyl groups on this bonding site hampers its reaction with other propagating species and the formation of a copolymer or network structure will be dependent on the size of the pendent group in the vinyl monomer.
Keywords: Degradation, hindered amine, photooxidation, polymeric HALS, stabilizers, synthesis.
Introduction
One of the main disadvantages of polymeric materials in relation to other classes of materials is their low stability to ultraviolet (UV) radiation. The degradation suffered by these materials when exposed to environmental conditions can range from mere surface discoloration affecting the aesthetic appeal of a product to extensive losses of mechanical properties, which severely limit their performance[1,2].
There are many pathways to increase the polymer resistance to the damage caused by the exposure to weathering. The blend of HALS derivatives from 2,2,6,6-tetramethylpiperidine into polymers in the molten state is among the most efficient pathways to stabilize polymers against ultraviolet radiation[3,4]. A great deal of effort has been spent on the understanding of the mechanisms involved in the stabilization of polymers by HALS. The scavenging of alkyl and peroxy radicals formed during polymer degradation by nitroxyl radicals is considered the usual mechanism by which HALS inhibits polymer oxidation. The structure of these amines is altered when exposed to UV radiation and in the presence of oxygen, producing nitroxyl radicals[5-10]. However the HALS stabilization mechanisms can be more complex and involve the quenching of excited polymer-oxygen complexes by HALS-oxygen and the formation of charge-transfer complexes (CTC) between peroxy radicals and HALS[11-14].
Regardless of the photostabilization mechanism assumed, the regeneration of the nitroxyl radicals is not complete and is responsible, in part, for the decrease in the HALS efficiency and deterioration in the polymer properties. Physical loss is another factor which contributes to reduce the photostabilizers efficiency. The ease of volatilization and poor resistance to extraction, due to a low compatibility between the polymer matrix and hindered amines, are the main phenomena involved in the physical loss of stabilizers[15-17]. Besides substantially reducing the HALS efficiency and therefore the product's lifespan, physical loss can cause serious toxicity problems. The synthesis of polymeric HALS, with optimal molecular weight and molecular architecture more compatible with different polymeric matrix has been an important alternative to achieve a long-term stabilizing effect and minimize the physical loss of HALS[18-20]. Polymeric HALS can be prepared by the copolymerization of vinyl monomers containing HALS group or macromolecular functional reaction[21,22]. Alternatively, many polymeric HALS can be prepared through the graft modification of preformed polymers[18].
This study investigated the functionalization of 2,2,6,6-tetramethylpiperidine derivatives and their posterior radical chain polymerization with vinyl monomers. 1-Acryloyl-2,2,6,6-tetramethylpiperidine (1ATP) and 4-acryloyloxy-2,2,6,6-tetramethylpiperidine (4ATP) were synthesized and subjected to radical chain polymerization with Sty and VAc to yield polymeric HALS. Thus, in the polymeric HALS produced, the structures derivatives from 2,2,6,6-tetramethylpiperidine are the active segment, responsible for the photostabilization effect, and the vinyl units are the inactive segment present to increase the molecular weight, reduce the diffusion rate, improve the compatibility with the polymeric matrix and reduce its physical loss. BTN was used as a chain-transfer agent (CTA) to achieve the optimal molecular weight of the synthesized copolymers.
Experimental
Materials
2,2,6,6-Tetramethyl-4-piperidinol (Aldrich, 98%), acryloyl chloride (Aldrich, 96%), methyl acrylate (Aldrich, 99%), BTN (Aldrich, 99%) and titanium (IV) isopropoxide (Aldrich, 97%) were used as received. 2,2,6,6-Tetramethylpiperidine (Merck, 99%) and triethylamine (Aldrich, 99%) were dried with anhydrous potassium carbonate (Carlo Erba, 99%) before use. Styrene (Aldrich, 99%) and vinyl acetate (Aldrich, 98%) were used after distillation. Azo-bis-isobutyronitrile (AIBN) used as a thermal decomposition initiator was kindly donated by Bayer from Brazil.
Synthesis of 1ATP and 4ATP monomers
1-Acryloyl-2,2,6,6-tetramethylpiperidine (1ATP) was prepared by a method similar to that used by Al-Malaika et al.[23]. In a three-neck flask reactor, 2,2,6,6-tetramethylpiperidine (21.3 mL; 0.13 mol) and triethylamine (17.5 mL; 0.13 mol) were mixed in 250 mL of dry benzene and maintained under stirring in ice-water bath. Acryloyl chloride (10.3 mL; 0.13 mol) was dissolved in 200 mL of dry toluene and added dropwise in the piperidine solution. The mixture was kept under stirring for 1 hour in ice-water bath and 8 hours at room temperature. The precipitate was removed by filtration and the liquid was washed with an aqueous sodium bicarbonate solution. The organic phase was separated and dried over anhydrous calcium carbonate. Active carbon was added to clear the solution and then removed by filtration. The solvents were removed under reduced pressure and the concentrate dissolved in hexane to precipitate the impurities. After the filtration and removal of hexane under reduced pressure, a yellow oil-like product (1ATP) was obtained.
4-Acryloyloxy-2,2,6,6-tetramethylpiperidine (4ATP) was prepared by a procedure described in the literature[24]. 2,2,6,6-Tetramethyl-4-piperidinol (15.69 g; 0.1 mol), methyl acrylate (8.4 mL; 0.1 mol) and titanium (IV) isopropoxide (3.0 mL) were added in a 250 mL flask charged with 100 mL of dry benzene. After stirring under nitrogen atmosphere for 1 h at ambient temperature, the mixture was heated to reflux temperature for 24 h and then repeatedly washed with an aqueous sodium bicarbonate solution after cooling. Benzene was removed under reduced pressure. The solid obtained was re-crystallized from chloroform, dried for several days under vacuum and stored in a dissecator.
Synthesis of 1ATP/Sty, 4ATP/Sty, 1ATP/VAc and 4ATP/VAc copolymers
The radical chain polymerization from hindered amines and vinyl monomers was carried out in toluene as a reaction medium. The AIBN concentration was 1.0 wt%, the molar ratio of hindered amine to vinyl monomers was 1:2, and the mass ratio of solvent/monomers mixtures was 2.5:1. The radical polymerization was performed in a 250 mL flask containing the above-mentioned proportions of comonomers and toluene, under nitrogen atmosphere. Various BTN concentrations were used to reduce the molecular weight of the copolymers. The 1ATP/Sty and 4ATP/Sty were polymerized for 10 h at 80ºC. The copolymers were precipitated in excess of methanol and purified by the reprecipitation from chloroform and methanol. The 1ATP/VAc and 4ATP/VAc polymerizations were carried out for 10 hours at 60ºC and the copolymers were reprecipitated in petroleum ether, collected and dried under vacuum at 40ºC for 8 h.
Characterization of monomers and copolymers
Infrared Spectroscopy (FTIR)
The structural modifications of 2,2,6,6-tetramethylpiperidine and 2,2,6,6-tetramethyl-4-piperidinol and the copolymers synthesized were characterized by infrared spectroscopy (FTIR) using a Spectrum 1000 Spectrophotometer from Perkin-Elmer, 4000-400 cm1 wavenumber range, with 24 scans for each spectra and resolution of 2 cm1. The samples were dissolved in chloroform, cast over potassium bromide (KBr) windows and dried under an infrared lamp.
Size Exclusion Chromatography (SEC)
The molecular weight distribution curves of the copolymers were determined by size exclusion chromatography (SEC) on a Waters 510 Chromatograph with a refractive index detector using three Ultrastyragel linear columns. The calibration curves were plotted using twelve samples of monodisperse polystyrene standards, ranging from 480 to 1×106 g mol1. Tetrahydrofuran was used as carrier at a flow rate of 1.0 ml min1 at room temperature. The average molar masses were determined by the Milennium 2010 software.
Carbon-13 Nuclear Magnetic Resonance Spectroscopy (13C NMR)
Carbon-13 NMR (13C NMR) spectra of monomers and copolymers were recorded on a Varian Unityplus 400 spectrometer, using magnetic field of 9.4T, pulse at 90º, 2,500 repetitions and relaxation time of 15 seconds to allow quantitative measurements. All samples were dissolved in chloroform in 5.0 mL probes and the carbon nuclei were observed at 100.57MHz. The chemical composition of the hindered amines and vinyl monomers was estimated from the quantitative analysis of their carbon-13 NMR spectra. The molar fraction of the hindered amines monomers is given by:

being FATP the mole fraction of the hindered amines (1ATP or 4ATP) in the copolymer, IATP and IVM the total integration of the signals of ATP and vinyl monomers (VM) carbon nuclei, respectively, and nATP and nVM the total number of the nuclei considered in each monomer.
The chemical composition of the copolymers was determined by:

being
w the weight-average molecular weight determined by SEC, x and y the molar ratio of the vinyl monomers and ATP in the copolymer, determined from Equation 1, respectively, A the molecular weight of the vinyl monomers (in g mol1) and B the molecular weight of ATP (in g mol1).Results and Discussion
Synthesis and characterization of 1ATP and 4ATP monomers
The reaction mechanism to yield 1ATP begins with the nucleophilic attack of the nitrogen in piperidine groups on the carbon of the carbonyl group in the acryloyl chloride. This reaction results in 1ATP and hydrochloric acid (HCl) as a by-product. The removal of the HCl from the reaction medium is important because it competes with acryloyl chloride to react with piperidine, a Lewis base, reducing the yield of the 1ATP. The strategy chosen to remove the HCl was to add triethylamine in the medium, a tertiary amine with pKa higher than the one of the piperidine. Thus the HCl reacts with tryethylamine to produce triethylamine hydrochloride, a precipitate with a slightly yellowish color, which is removed by filtration at the end of the reaction. Scheme 1 shows the 1ATP synthesis in a simplified manner.
The FTIR spectra of 2,2,6,6-tetramethylpiperidine and 1ATP are shown in Figure 1. Secondary amines, as piperidines, show a very narrow peak in the 3400-3100 cm1 region, relative to the N-H stretch (Figure 1a). 1ATP spectrum shows no absorption in this region, a clear indication that the amine reacted completely with the acryloyl chloride (Figure 1b). The absorptions slightly below 3000 cm1 are assigned to the saturated C-H stretch (methyl and methylene groups) and unsaturation is responsible for the absorptions above 3000 cm1: C-H antisymmetric stretch at 3080 cm1 and C-H symmetric stretch at 3010 cm1. Unsaturation also absorbs at 1604 cm1 due to C=C stretch and is indispensable to subsequent radical polymerizations with other vinyl monomers. In the reaction between hindered amine and acryloyl chloride, the amide is formed and this new group presents a strong absorption at 1645 cm1 due to the stretch of the C=O bond.
The 1ATP was also characterized by carbon-13 NMR and its spectrum in CDCl3 is shown in Figure 2. The signal at 30.0 ppm corresponds to four methyl groups (CA5). Peaks at 14.2 and 36.1 ppm can be ascribed to the methylene groups (CA4 and CA3) in different positions of the piperidine ring. The chemical shift at 169.6 ppm has been assigned to the carbon (CA1') of the amide group, confirming the synthesis of 1ATP. Vinyl unsaturated carbons (CA2' and CA3') are in δ 120-140 ppm. Unreacted piperidine was hardly seen by carbon-13 NMR which is corroborated by the absence of absorption at 3300 cm1 in the 1ATP infrared spectrum indicating that the amine reacted completely.
In the synthesis of 4ATP (scheme 2), titanium (IV) isopropoxide (Ti(OPr)4) was used as a Lewis acid catalyst to favor the reaction between the methyl acrylate and the hydroxyl group and avoid the reaction with amine group in the piperidinol, although the amine is more reactive than hydroxyl. The transesterification reaction of the synthesis of 4ATP begins by the formation of a coordination bond between the oxygen of the hydroxyl group in the piperidinol and the metallic center of the titanium catalyst, followed by the nucleophilic attack to the carbonyl group of the methyl acrylate, resulting in ester (4ATP) and methanol. Transesterification reaction is an equilibrium reaction which can be shifted in either direction by the control of the reaction medium. The use of reagents in stoichiometric amounts allowed the reaction to continue until it had reached the equilibrium, explaining the low yield of the 4ATP, i.e., 32%.
The FTIR spectrum of 2,2,6,6-tetramethyl-4-piperidinol (Figure 3a) shows a broad absorption in the 3600-3100 cm1 range due to the O-H stretch overlapped by a narrow absorption relative to the N-H stretch at 3255 cm1. The analysis of 4ATP spectrum (Figure 3b) shows that the broad absorption of the hydroxyl at 3300 cm1 is completely absent, while the narrow absorption due to the presence of amine is sharp, indicating that the experimental conditions favored the reaction between the methyl acrylate and the hydroxyl group of the piperidinol, as expected.
The carbonyl and vinyl unsaturation absorb at 1720 cm1 and 1604 cm1, respectively. The 4ATP spectrum still shows absorptions relative to the ester groups at 1295 cm1 assigned to the stretch of O-C(=O)-C and 1064 cm1 due to the stretch of C-O. The absorption relative to the C-O stretch is also present in the piperidinol spectrum. Although the absence of hydroxyl is evident in the 4ATP spectrum, the confirmation of the formation of an ester (4ATP) by FTIR is not granted because this group is also present in the chemical structure of methyl acrylate.
In the carbon-13 NMR spectra of 4ATP (Figure 4), the carbonyl carbon of the ester group (CA1') shows a signal at δ=166.0 ppm. The signal at δ=69.3 ppm can be assigned to the carbon in the piperidine ring, nearest the ester group (CA4). The chemical shift at 35.7 ppm is assigned to the four carbons of the methyl group (CA5) while carbons of the vinyl unsaturation (CA2' and CA3') show signals in the 125-135 ppm range.
Synthesis and characterization of 1ATP/Sty, 4ATP/Sty, 1ATP/VAc and 4ATP/VAc copolymers
In the radical polymerization of 1ATP and Sty the
wof the copolymer formed was 63,000 g mol1, which is considered high even for a polymeric HALS. In this type of polymerization, the use of a chain-transfer agent (CTA) is viable to control the final molecular weight of the polymer. In choosing a CTA, two parameters must be considered: transfer constants (CS) of the CTA and their concentration. In radical polymerization with Sty at 60ºC, the value of CS for thiols like BTN is 21.00[25], highest for conventional transfer-agents. Table 1 summarizes the results of the polymerizations of 1ATP with Sty under various BTN concentrations.
The maximum molecular weight reduction for the 1ATP/Sty copolymerization was obtained with BTN concentration of 31.9 mM. This is a 80%-reduction in the
w when compared with the value obtained under the same conditions, but without BTN. The increase in the BTN concentration also results in decreased polydispersity, which is a parameter that measures the width of molecular weight distribution curves calculated by ratio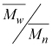 . No precipitate could be obtained even increasing the BTN concentration up to 160 mM, indicating that the partial scavenging of the radical species occurs soon after the opening of the double bond. Therefore the product should have a low molecular weight, close to that of the monomers. The copolymer obtained with 31.9 mM of BTN showed
. No precipitate could be obtained even increasing the BTN concentration up to 160 mM, indicating that the partial scavenging of the radical species occurs soon after the opening of the double bond. Therefore the product should have a low molecular weight, close to that of the monomers. The copolymer obtained with 31.9 mM of BTN showed 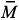 w of 12,800 g/mol, and its carbon-13 NMR spectrum is depicted in Figure 5.
w of 12,800 g/mol, and its carbon-13 NMR spectrum is depicted in Figure 5.
The analysis of this NMR spectrum allows one to state that the magnitude of the signals related to the Sty carbons is more intense than that of 1ATP. The shifting of the signal of the carbonyl carbon of the amide group (CA1') to δ=180.9 ppm indicates an opening in the double bond during the radical polymerization. The mole fraction of 1ATP and Sty in the copolymer was determined by the ratio between the area of each sign and the number of nuclei responsible for the signal. Integrating the signals at 14.0 ppm (CA4) and 127.5 ppm (CB2), the molar ratio of 1ATP:Sty in the copolymer synthesized is 1:6.5. Knowing that the molecular weights of Sty and 1ATP are 104.1 g mol1 and 195.3 g mol1, respectively, and following equations (1) and (2), we can estimate that this copolymer is made by 95 units of Sty and 15 units of 1ATP. The mole fraction of 1ATP:Sty determined in the copolymer, 1:6.5, is very different from that used in the feed, 1:2.0, meaning that the Sty reactivity is higher than that of 1ATP for the synthesis conditions employed in this work.
In the copolymerization of 1ATP and VAc without the use of BTN, the polymer presented a of 108,000 g mol1. Using a BTN concentration of 0.30 M molecular weight is reduced to 28,000 g mol1. BTN concentrations above 0.62 M inhibits the polymerization altogether.
The FTIR spectrum of the polymer synthesized with BTN concentration of 0.30 M shows that in the region between 1870-1540 cm1, characteristic of carbonyl-containing functional groups, the absorption at 1645 cm1 is due to the carbonyl of the amide group present in 1ATP. However, in this region the FTIR spectrum does not show absorption corresponding to the carbonyl of the ester group present in VAc. Moreover, the absence of additional absorptions at 1240 cm1 and 1050 cm1, characteristic of the ester group, indicates that the polymer synthesized does not present VAc units in its chemical structure. The characterization of this polymer by carbon-13 NMR shows a single signal at δ=178.7 ppm, which is a characteristic region of carbonyl carbons of the amide group. The analyses of this polymer by FTIR and 13C NMR allow concluding that the polymeric chain is constituted only by 1ATP units, i.e., poly(1ATP) homopolymer.
The radical polymerization of 4ATP and vinyl monomers showed a different behavior. In the 4ATP/Sty copolymerization using molar ratio 4ATP:Sty of 1:2, the SEC measurement confirmed that its molecular mass was below 480 gmol1. Increasing molar ratio 4ATP:Sty to 1:4, a copolymer was formed. Figure 6 depicts its infrared spectrum. This spectrum shows that the copolymer is made by styrenic and 4ATP units. The absorptions at 3060 cm1 and 3025 cm1 correspond to the aromatic or unsaturated C-H stretch and the absorption at 1605 cm1 is due to the symmetric ring stretch. Both stretches are characteristic of the styrenic unit. The presence of the 4ATP units in the copolymer is confirmed by the absorptions at 1724 cm1, which corresponds to the axial deformation of carbonyl of ester group, and at 1245 cm1 and 1050 cm1, corresponding to the stretching of the ester groups. However an outstanding characteristic of this spectrum is the absence of amine absorption at 3320 cm1, indicating that the hydrogen atom of the amine group reacted with radical species present in the medium, even in the absence of oxygen. Under these conditions the hydrogen atom is sufficiently labile to react with present radical species and terminate prematurely the growth of the chains. For this competitive scavenging mechanism, the 4ATP acts as a CTA, which explains the difficulty to obtain a polymer when 4ATP is used as a monomer.
4ATP/Sty copolymer was characterized by 13C NMR and the values of the integrals of the signals indicate that the molar ratio 4ATP:Sty in the copolymer is 1:5.5, similarly to the value of the molar ratio used in the feed. The SEC results have shown that the molecular weight distribution curve of 4ATP/Sty copolymer is monomodal with wof 11,000 g mol1 and polydispersity of 1.25. Knowing that the molecular weights of 4ATP and Sty are 211.30 g mol1 and 104.15 g mol1, respectively, the 4AT/Sty copolymer is made by 14 units of 4ATP and 77 units of Sty.
A similar behavior was observed in the copolymerization of 4ATP with VAc. However to get a polymer the amount of VAc had to be increased up to a feeding molar ratio of 1:6. Differently of the 4ATP/Sty copolymer, this polymer was insoluble in several organic solvents, indicating the formation of a crosslinked structure.
The difference in behavior when 4ATP was used as one of the comonomers can be attributed to its reactivity and hindrance. Besides opening the p-bond to form a new radical, the N-H bond is relatively less stable under the conditions employed and therefore able to generate radicals by homolytic bond breakage. The presence of hydrogen radicals explains the difficulty to obtain a copolymer in the synthesis with 4ATP. A high 4ATP:vinyl monomers ratio, such as 1:2 results in high concentrations of hydrogen radicals which react prematurely with propagating species and prevent the growth of the chain.
The high hindrance imposed by the four methyl groups on the radical formed in the nitrogen atom inhibits the formation of a new chemical bond with monomers whose pendant group has a large volume, as the case of Sty. The difficulty of establishing an effective chemical bond between the nitrogen and the Sty results in the formation of a soluble copolymer. However the pendant group in VAc would not be large enough to avoid the formation of an effective chemical bond with the radical generated in the nitrogen atom, forming the crosslinked structure.
Conclusions
The structural modifications of 2,2,6,6-tetramethylpiperidine using acryloyl chloride and 2,2,6,6-tetramethyl-4-piperidinol using methyl acrylate have been successfully accomplished, producing two new structures (1ATP and 4ATP) able to be polymerized with vinyl monomers by free-radical reactions. The use of various BTN concentrations resulted in a significant decrease in the molecular weight of 1ATP and vinyl copolymers.
In the presence of radicals and a reaction medium without oxygen, the hydrogen atom bounded to the nitrogen has proved to be labile enough to react with propagating radicals. Due to this characteristic, the 4ATP acts as a chain-transfer agent. The volume of the pendant group in vinyl monomers is important in order to get a soluble copolymer, because the radical formed in the nitrogen atom presents high hindrance imposed by the neighboring methyl groups. Therefore the copolymerization of 4ATP and Sty produced soluble products, while 4ATP and VAc presented a network structure.
Acknowledgements
The authors would like to acknowledge National Council for Scientific and Technological Development (CNPq) for the financial support provided to this research.
Received: 10/06/13
Accepted: 02/24/14
- 1. Andrady, A. L.; Hamid, S. H.; Hu, X. & Torikai, A. J. Photochem. Photobiol. B: Biol., 46, p.96 (1988). http://dx.doi.org/10.1016/S1011-1344(98)00188-2
- 2. Feldman, D. J. Polym. Environ., 10, p.163 (2002). http://dx.doi.org/10.1023/A:1021148205366
- 3. Rabek, J. F. "Polymer Photodegradation: Mechanisms and Experimental Methods", Chapman & Hall, London (1995).
- 4. Ammala, A.; Bateman, S.; Dean, K.; Petinakis, E.; Sangwan, P.; Wong, S.; Yuan, Q.; Yu, L.; Patrick, C. & Leong, K. H. Prog. Polym. Sci., 36, p.1015 (2011). http://dx.doi.org/10.1016/j.progpolymsci.2010.12.002
- 5. Pilař, J.; Michálková, D.; Šedĕnková, I.; Pfleger, J. & Pospíšil, J. Polym. Degrad. Stab., 96, p.847 (2011). http://dx.doi.org/10.1016/j.polymdegradstab.2011.02.004
- 6. Rossi, I.; Venturini, A. & Zedda, A. J. Am. Chem. Soc., 121, 7914 (1999). http://dx.doi.org/10.1021/ja9906655
- 7. BilIingham, N. C. & Calvert, P. D. "The Degradation and Stabilisation of Polyolefins An Introduction", in: Degradation and Stabilisation of Polyolefins, cap. 1, Norman S. Allen (ed), Applied Science, London (1983).
- 8. Carlsson, D. J. & Wiles, D. M. J. Macromol. Sci. C: Polym. Rev., 14, p.65 (1976). http://dx.doi.org/10.1080/15321797608076113
- 9. Delprat, P.; Duteurtre, X. & Gardette, J. L. Polym. Degrad. Stab., 50, p.1 (1995). http://dx.doi.org/10.1016/0141-3910(95)00061-P
- 10. Philippart, J. L.; Posada, F. & Gardette, J. L. Polym. Degrad. Stab., 49, p.285 (1995). http://dx.doi.org/10.1016/0141-3910(95)87010-5
- 11. Gugumus, F. Polym. Degrad. Stab., 40, p.167 (1993). http://dx.doi.org/10.1016/0141-3910(93)90208-Z
- 12. Gijsman, P. Polym., 43, p.1573 (2002). http://dx.doi.org/10.1016/S0032-3861(01)00708-X
- 13. Gugumus, F. Die Angew. Makromol. Chem., 176, p.241 (1990). http://dx.doi.org/10.1002/apmc.1990.051760121
- 14. Gugumus, F. Polym. Degrad. Stab., 39, p.117-135 (1993). http://dx.doi.org/10.1016/0141-3910(93)90131-2
- 15. Minagawa, M. Polym. Degrad. Stab., 25, p.121-141 (1989). http://dx.doi.org/10.1016/S0141-3910(89)81004-3
- 16. Smoliak, L. Y. & Prokopchuk, N. R. Polym. Degrad. Stab., 82, p.169 (2003). http://dx.doi.org/10.1016/S0141-3910(03)00180-0
- 17. Malík, J.; Hrivík, A. & Alexyová, D. Polym. Degrad. Stab., 35, p.125 (1992). http://dx.doi.org/10.1016/0141-3910(92)90103-C
- 18. Xue, B. & Toyota, A. Polym. Bull., 62, p.327 (2009). http://dx.doi.org/10.1007/s00289-008-0028-5
- 19. Sun, G. J.; Jang, H. J.; Kaang, S. & Chae, K. H. Polym., 43, p.5855 (2002). http://dx.doi.org/10.1016/S0032-3861(02)00514-1
- 20. Singh, R. P.; Patwa, A. N.; Desai, S. M.; Pandey, J. K.; Solanky, S. S. & Prasad, A. V. J. Appl. Polym. Sci., 90, p.1126 (2003). http://dx.doi.org/10.1002/app.12672
- 21. Pan, J.; Cheng, W.; Song, Y. & Hu, X. Polym. Degrad. Stab., 39, p.85 (1993). http://dx.doi.org/10.1016/0141-3910(93)90128-6
- 22. Pan, J. Q. Chin. J. Polym. Sci., 19, p.341 (2001).
- 23. Al-Malaika, S.; Ibrahim, A. Q. & Scott, G. Polym. Degrad. Stab., 22, p.233 (1988). http://dx.doi.org/10.1016/0141-3910(88)90013-4
- 24. Al-Malaika, S.; Scott, G. & Wirjosentono, B. Polym. Degrad. Stab., 40, p.233 (1993). http://dx.doi.org/10.1016/0141-3910(93)90210-A
- 25. Odian, G. "Principles of Polymerization", John Wiley, New Jersey (2004).
Publication Dates
-
Publication in this collection
03 Apr 2014 -
Date of issue
2014
History
-
Received
06 Oct 2013 -
Accepted
24 Feb 2014









