Abstract
The graft copolymer with semi-interpenetrating polymer networks (semi-IPNs) from 2-hydroxyethyl methacrylate (HEMA) and natural rubber (NR) latex was prepared using cumene hydroperoxide and tetraethylene pentamine redox initiator system. The changes of grafting ratios and grafting efficiency with the reaction time and temperature, the concentration of crosslinking agent, initiator and monomer were investigated. The appropriate amounts of crosslinking agent (0.1phr), initiator (0.2phr) and monomer (20phr) and the optimum reaction conditions of 16ºC×8h were determined. The swelling temperature and times of monomers against NR latex particles were found to be significant for the grafting copolymerization and the appropriate swelling conditions were 16ºC×20h. The water contact angle measurement and platelet adhesion evaluation indicated that the hydrophilicity and blood compatibility of NR latex could be improved by grafting copolymerization with HEMA.
Natural rubber latex; graft copolymerization; 2-hydroxyethyl methacrylate; redox initiator system; hydrophilicity and blood compatibility
Preparation and Structural Characterization of Hydroxylethyl Methacrylate Grafted Natural Rubber Latex
Feiyun WeiI; Heping YuI,IV; Zongqiang ZengI; Hongchao LiuI; Qifang WangI; Jianlong WangII; Sidong LiIII
IAgriculture Ministry Key Laboratory of Tropical Crop Products Processing, Agricultural Product Processing Research Institute, Chinese Academy of Tropical Agricultural Sciences, China
IISchool of Chemical Engineering and Environment, North University of China, China
IIIFaculty of Science, Guangdong Ocean University, China
IVAgriculture Ministry Key Laboratory of Tropical Crop Products Processing, Agricultural Product Processing Research Institute, Chinese Academy of Tropical Agricultural Sciences, NO.48, South Renmin Road, Zhanjiang 524001, Guangdong, China, e-mail: wfydear@163.com
ABSTRACT
The graft copolymer with semi-interpenetrating polymer networks (semi-IPNs) from 2-hydroxyethyl methacrylate (HEMA) and natural rubber (NR) latex was prepared using cumene hydroperoxide and tetraethylene pentamine redox initiator system. The changes of grafting ratios and grafting efficiency with the reaction time and temperature, the concentration of crosslinking agent, initiator and monomer were investigated. The appropriate amounts of crosslinking agent (0.1phr), initiator (0.2phr) and monomer (20phr) and the optimum reaction conditions of 16ºC×8h were determined. The swelling temperature and times of monomers against NR latex particles were found to be significant for the grafting copolymerization and the appropriate swelling conditions were 16ºC×20h. The water contact angle measurement and platelet adhesion evaluation indicated that the hydrophilicity and blood compatibility of NR latex could be improved by grafting copolymerization with HEMA.
Keywords: Natural rubber latex, graft copolymerization, 2-hydroxyethyl methacrylate, redox initiator system, hydrophilicity and blood compatibility
Introduction
The chemical modification is a main way to endow natural rubber (NR) with new physical and chemical properties and thus extends its applications. To date, many methods such as substitution[1], addition[2,3], cyclo-addition[4,5], electro-cyclic reaction and the "ene"[6] reaction, have been attempted to prepare a series of NR derivatives such as hydrogenated NR, epoxidized NR, chlorinated NR, cyclised NR and graft copolymerized NR. These modifications have not only been directed towards the enhancement of certain characteristic properties of NR, but also to introduce totally new properties not usually associated with it.
Among these derivatives from NR, the graft copolymerization of vinyl monomers towards NR is always an important research direction, because this method not only maintains the intrinsic properties of NR, but also exhibits the characteristic properties of the homopolymer formed from the monomer being used. In fact, only the commercialized productions of graft copolymerization of vinyl monomers with NR latex have been achieved till today. During the past century, quite a lot of monomers have been investigated to graft copolymerized with NR in latex form directly or in solutions. These monomers include methyl methacrylate[7-11], styrene[12-15], vinyl acetate, acrylonitrile[16,17], acrylamide, methyl acrylamide[18], dimethylaminoethyl methacrylate[19,20], 2-hydroxyethyl methacrylate(HEMA)[21],fluoro-alkylethyl metracrylate[22] and so on. Considering the benefit to the environment, the graft copolymerization conducted directly in NR latex is more available. And the NR latex seeded emulsion polymerizations have been investigated extensively. However, it seems only the NR latex composites prepared from the graft copolymerization of methyl methacrylate and styrene have been successfully used as adhesives, paints and toughening agents for thermoplastics.
As the NR latex is a typical renewable resource and exhibits excellent film-forming ability, resilience, flexibility and wet-gel strength, it is still the dominated material for the productions of medical devices such as medical gloves, catheters and condoms even if the synthetic polymers such as silicone rubber, polyethylene, polytetrafluoroethylene and polyvinyl chloride are also the available ones. However, although the NR itself has good biocompatibility, the non-rubber components in NR latex greatly influence the utilizing safety of those medical products made from NR latex. In aim to eliminate the influences of non-rubber components, many methods have been tried to reduce the non-rubber contents. However, as the non-rubber components are the key matters on the colloid stability and technological behaviour of latex, the decrement in non-rubber contents will affect the colloid stability and technological behaviour of NR latex. In recent decades, the surface modifications of NR latex films through the graft copolymerization of vinyl monomers[23-26] and the immobilization of phospholipid[27] have been conducted to improve the biocompatibility of medical devices. The coatings formed onto the surfaces of NR latex films are beneficial to prevent the non-rubber components and residual compounding agents from diffusing towards the surfaces of NR latex films. In addition, some of the coatings are of good biocompatibility. However, the difference of mechanical properties between the immobilized coatings and rubber matrix will reduce the utilizing life of the products. And the irregular shape of medical devices will also lead to un-homogeneous coatings. Therefore, the improvement of the biocompatibility of medical device made from NR latex is still a challenge problem.
As the monomer HEMA is well known for the excellent hydrophilicity and biocompatibility and the poly(HEMA) also exhibits outstanding tissue compatibility due to the similarity of physical properties to human tissues, the NR composite latex is expected to be improved in hydrophilicity and biocompatibility by grafting copolymerization with HEMA. This will be an available method to improve the biocompatibility of medical devices from raw materials.
Recently, the graft copolymer of natural rubber (NR) and 2-hydroxyethyl methacrylate (HEMA) was prepared by adding 5 phr of HEMA with 0.15mol% of initiator at 60ºC×90 min[21]. As reported, the grafting ratio was only about 8.35%. Because the water soluble HEMA intends to partition in serum especially in high temperatures and exhibits low swelling efficiency against NR latex particles, it is very difficult to achieve the graft copolymerization of HEMA with NR latex particles. To improve the grafting ratio by increasing the monomer dosage, the colloid stability of latex composite would be reduced under a routine technology of grafting copolymerization. Furthermore, only the peeling strength was investigated when the latex composite was used as adhesive in this paper. In this work, the graft copolymerization of HEMA with NR latex was investigated to increase the grafting ratio by improving the reaction conditions. And the hydrophilicity and anti-coagulation behaviour against blood were also evaluated.
Experimental
Materials
NR latex used was commercial high-ammonia NR latex concentrate with a dry rubber content (DRC) of 60wt%. The monomer (HEMA, purity of 99wt%) with hydroquinone monomethyl ether (MEHQ) as inhibitor was purchased from Tokyo Kasei Kogyo Co. Ltd., Japan. The cumene hydroperoxide (CHPO, purity of 80wt%), tetraethylene-pentamine (TEPA, purity of 80wt%) and tetraethylene-glycol dimethacrylate (TEGDMA, purity of 99wt%) were purchased from Aldrich.
Preparation of NR-g-HEMA
The graft copolymerization of HEMA and NR latex was conducted in a 500mL glass reactor through seeded emulsion polymerization. NR latex (100g) was charged into the reactor and then the HEMA, CHPO and TEGDMA were added successively by stirring. NR latex was swollen with the HEMA for 24h at the corresponding reaction temperature before the TEPA was added. The mixture was diluted to a total solid content of 20wt% with distilled water and then allowed to react at 10-20ºC for the time required. After the completion of the reaction, the latex was coagulated with 5wt% acetic acid and the coagulum was washed with distilled water at room temperature for 24h to remove the un-grafted monomers and then was subjected to Soxhlet extraction using ethanol for extracting un-grafted homopolymers poly(HEMA). The gross copolymer was recovered and dried to constant mass in an oven at 70ºC. The variable factors for the graft copolymerization are shown in Table 1. The grafting ratio (G) and grafting efficiency (GE) were calculated using the following relationships:

Where W0 is the dry weight of NR latex, W and W1 are the weights of the dried sample before and after extraction, respectively.
The NR-g-HEMA latex was cast at room temperature in an open glass tray to obtain a film with final thickness of ca.1.5mm. Once dried naturally, the film was rinsed with distilled water for 24h to remove the un-grafted HEMA monomers and subjected to Soxhlet extraction for 24h to remove un-grafted HEMA homopolymer. Then the film was air-dried for the subsequent analyses.
FTIR analyses
The ATR-FTIR analyses of NR-g-HEMA latex film surfaces were performed on a TENSOR 27 spectrometer using a ZnSe crystal with multi-reflection mode. The scanning frequency is 16 and the spectral resolution is 4 cm1. The incident angle is 45º. The spectra were recorded in the range of 4000 cm1 to 600 cm1.
Measurements of static water contact angles and water absorption ratio
The measurements of static water contact angles of NR-g-HEMA latex films were performed on a SL200B telescopic goniometer at 25±1ºC and 50%±5% relative humidity. Meanwhile, the surface energy of NR-g-HEMA latex films was also calculated using the following equation:

where, γLand γS are surface energy of distilled water and NR-g-HEMA latex films respectively, and γL is set to72 mN/m. θ is the contact angle and β is a constant and is set to 0.0001247 (mN/m)2 according to Ref [28].
The NR-g-HEMA latex films with known weights were immersed in distilled water for 48h at ambient temperature (25ºC). Then the swollen films were removed and blotted dry between two pieces of filter paper before weighing. The water absorption ratio (S) was calculated as:

where W0 and W1 are the weights of sample before and after immersion in water, respectively.
Determination of particle sizes and distributions
The particle sizes and distributions of NR-g-HEMA latex were determined on a Malvern Master Sizer 2000 and the particle sizes from 0.02 to 20µm were recorded.
Evaluation of platelet adhesion
The films of NR latex and NR-g-HEMA latex with grafting ratio of 14.0% were washed with distilled water and PBS (pH=7.4) and contacted with freshly concentrated human platelet-rich plasma (PRP) at 37ºC for 2h. Then the films were rinsed with PBS to remove un-adhered platelets and treated with 2.5% glutaraldehyde for 1h at ambient temperature. After washed with PBS, the films were dehydrated by immersing systematically in a series ethnol-water solutions (50, 60, 70, 80, 90, 100 in v/v) for 30min each and allowed to evaporated at room temperature. The platelet attached surfaces were observed on a Philips XL30 scanning electronic microscope (SEM) after having gold-coated for 2 minutes in an Hitachi E-1010 gold sputter (Hitachi, Japan).
Results and Discussion
Swelling of NR latex with HEMA
It is well known that the vinyl monomers have to contact with and swell NR latex particles sufficiently before the graft copolymerization proceeds. As NR latex is a colloid with NR latex particles (rubber phase) dispersing into serum (water phase), the oil-soluble monomers such as methyl methacrylate (MMA) and styrene (St) intend to partition in rubber phase and swell NR latex particles sufficiently in 1h at room temperature. Thus, the graft copolymerization of MMA and St with NR latex initiated by a redox initiating system can be carried out easily. However, the water soluble HEMA intends to partition in serum and exhibits low swelling efficiency against NR latex particles. Therefore, it is very difficult to achieve the graft copolymerization of HEMA with NR latex particles. In this work, it was tried to increase the swelling efficiency of HEMA against NR latex particles by reducing the swelling temperature and prolonging the swelling time. The results indicate that the NR latex particles could be swollen sufficiently for the subsequent graft copolymerization by reducing the temperature below 20ºC and prolonging the swelling time to 20h. However, NR latex could be clotted by freezing if the swelling temperature was lower than 10ºC. As the subsequent grafting copolymerization reaction could be conducted properly at about 16ºC, the swelling procedures of NR latex with HEMA were carried out at 16ºC in the following experiments. At this temperature, the NR latex should be swollen for 20h at least. Or else, there was nearly no HEMA having grafted onto NR molecules. The swelling times were further prolonged to 24h in our work actually to ensure the swelling efficiency.
Effect of reaction temperature and time
The reaction temperature imposes a strong effect on the grafting copolymerization of HEMA with NR latex. The HEMA can be polymerized with a redox initiator system rapidly even at room temperature, which will reduce the grafting ratio of HEAM onto NR molecules greatly. In addition, the formation of great amounts of homopolymers will reduce the stability of NR latex and even lead to the coagulation of NR latex because of the water hydrophilic and water absorbing performances of poly(HEMA). Reducing reaction temperature is beneficial to the partitioning of HEMA in rubber phase and thus the polymerization rate of HEAM to form homopolymers can be reduced.
In this work, the reaction temperature was reduced below 20ºC and the changes of G(%) and GE(%) with reaction temperature analyzed from 14ºC to 20ºC. As shown in Figure 1, the G(%) and GE(%) increased when the reaction temperature was risen from 14ºC to 16ºC and then decreased when the temperature was further risen to 20ºC. Therefore, the reaction temperature of 16ºC was selected.
The G(%) and GE(%) as a function of reaction time was investigated over 4 to 10h, by fixing the concentration of HEMA, CHPO, TEGDMA and TEPA at 20phr, 0.2phr, 0.1phr and 0.2phr by weight, respectively. NR latex was swollen with HEMA at 16ºC for 24h and subsequently reacted at 16ºC for 8h. The result is presented in Figure 2. It can be seen that the G(%) increased slightly by prolonging reaction time from 4h to 8h and increased to 14% by prolonging reaction time to 8h. Then the curve almost paralleled to time axis after 8h. And at the same time, the GE(%) decreased due to the increasing conversion of free homopolymers. Thus, the reaction time was determined as 8h in this work.
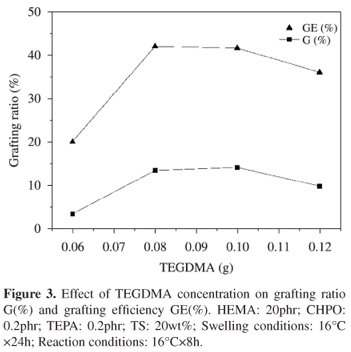
Effect of TEGDMA concentration
It seems impossible to conduct the graft copolymerization of HEMA with NR latex unless crosslinking agent TEGDMA was added into the latex. The effect of TEGDMA concentration on G(%) and GE(%) was investigated over the range of 0.06 to 0.12phr by keeping the concentration of all other reagents constant. The swelling and reaction conditions were 16ºC×24h and 16ºC×8h, respectively. From Figure 3 it can be seen that G(%) increased rapidly with TEGDMA level increasing from 0.06 to 0.1phr and then slightly decreased with TEGDMA amount increasing to 0.12phr.
TEGDMA is commonly used as a crosslinking agent to form the semi-interpenetrating polymer networks (semi-IPNs) which can be prepared by dissolving a preformed linear polymer in the mixture of hydrophilic monomer and cross-linking agent. In this way, a synthetic network was formed around primary polymer chains.
Effect of monomer concentration
The effect of monomer concentration on G(%) and GE(%) was investigated from 12 to 24 phr at the fixed concentration of other agents. The NR latex was swollen with HEMA at 16ºC for 24h and subsequently reacted at 16ºC for 8h. It can be seen from Figure 4 that G(%) increased with the increase in HEMA amount. But GE(%) was slightly decreased when the HEMA amount got to 24 phr.
It should be noted that the stability of NR latex would reduce dramatically when the HEMA amount was larger than 24phr. In addition, the film-forming performance of NR-g-HEMA latex was poorer than that of NR latex and decreased gradually with the increment of G(%), due to the hydrophilic property of poly(HEMA). In fact, the coagula yielded by coagulating NR-g-HEMA latex with acetic or formic acid were cohesionless particles, but could cohere together after dried. Thus, the proper amount of HEMA was selected as 20phr in this work.
Effect of initiator concentration
The effect of initiator concentration on G(%) and GE(%) was discussed over the range of 0.12 to 0.24 phr by keeping the amount of monomer and other agents constant. The result was presented in Figure 5. The value of G(%) increased obviously by increasing initiator level from 0.12phr to 0.2phr, and then the increasing trend became less significant by further increasing initiator concentration to 0.24phr. This can be explained by the mechanism of graft copolymerization which had been studied by many authors. CHPO can be decomposed by TEPA to form free radical whose function is to abstract α-H from NR molecules and thus produce active site on the backbone of NR molecules. The number of such active sites increases as the initiator amount increases. At a lower level of CHPO, the homopolymerization of HEMA was predominated over grafting due to the lower number of active sites, leading to a relatively low G(%). On the contrary, more active sites can be generated at a higher level of CHPO, thus the grafted copolymers were formed more readily than the homopolymers, leading to a higher G(%).
These features also can be confirmed by determining the particle sizes and distribution of NR-g-HEMA latex (Figure 6). It can be seen that the particle size distribution curve of NR latex exhibited a unimodal distribution pattern with particle sizes varying from 0.36µm to 1.66µm (curve-e in Figure 6), whereas those of modified latex were much wide and could be bimodal or unimodal depending on the CHPO level. When the CHPO amount was less than 0.2phr, the homopolymerization of HEMA was so obvious that the bimodal particle size distribution curves of modified latex (curve-a, curve-b and curve-c in Figure 6) could be formed. Only when the CHPO amount increased to 0.24phr, the unimodal particle-size distribution curve could be determined (curve-d in Figure 6). It is obvious that the grafting copolymerization can be promoted by increasing the CHPO amount.
In addition to the particle size distribution, the CHPO amount also exhibited influence on particle sizes of NR-g-HEMA latex. From Figure 6 it can be seen that the particle sizes of NR-g-HEMA latex decreased as the CHPO amount increased. The particle size depends on the chain length of poly(HEMA) grafted onto NR molecules. It is probable that the monomers were grafted onto NR molecules by propagations to form relatively long poly(HEMA) molecules at low level of CHPO. By increasing the initiator amount, more reactive sites could be formed on NR molecules and thus the relatively short poly(HEMA) molecules might graft onto NR latex molecules. As a result, the NR-g-HEMA latex particle sizes decreased slightly with the particle size distribution shifting towards that of NR latex. The increases of latex particle sizes further indicated that the HEMA had been grafted onto NR latex successfully.
FTIR-ATR analyses
The NR-g-HEMA molecular structure was characterized by using FTIR-ATR. As illustrated in Figure 7, the ATR-FTIR spectrum of NR-g-HEMA latex film was different from that of NR latex. The characteristic peaks of cis-1,4 polyisoprene, at 2960 cm1, 2927 cm1, 2852cm1 (CH stretching), 1661 cm1 (C=C stretching), 1448 cm1 (CH deformation of CH2), 1376 cm1 (CH deformation of CH3) and 835cm1 (CH deformation of C= CH), could be contributed to NR molecules[25,26]. In the spectrum of NR-g-HEMA latex film, however, in addition to absorption peaks of cis-1,4 polyisoprene, the absorption peak of C=O stretching of poly(HEMA) at 1722cm1 was observed obviously, indicating that the poly(HEMA) molecules had been introduced onto NR molecules successfully.
Effects of grafting ratio on water contact angle and water absorption ratio
The NR-g-HEMA latex films with different grafting ratios were prepared to determine the static water contact angles. As shown in Figure 8, the water contact angle of NR latex film was about 97º. After grafted with poly(HEMA), the water contact angle of NR-g-HEMA latex film decreased gradually with the increase of G(%) due to the hydrophilicity of poly(HEMA) and could be reduced to about 68º as the G(%) was up to 16.2%. Meanwhile, the surface energy was also estimated and the results were shown in Table 2. The surface energy almost linearly increased as the G(%) rising, which can be explained by the improvement of NR-g-HEMA latex film's polarity and hydrophilicity. The results clearly demonstrated that it is possible to improve the hydrophilicity of NR latex film through grafting copolymerization of with HEMA. This result was further confirmed by measuring the water absorption ratio S(%) of NR-g-HEMA latex film. As shown in Table 2, the S(%) of NR-g-HEMA latex film increased as the G(%) increased and could be as high as 22.93% when the G(%) of NR-g-HEMA latex was up to 16.2%, which was 6 times higher than that of NR latex film.
Platelet adhesion behaviour of NR-g-HEMA latex film
Figure 9 shows the SEM photos of platelet adhesions onto the films of NR latex and NR-g-HEMA latex. It can be seen that there were a large number of platelets adhered on the surface of NR latex film, and some of platelets were distorted with pseudopodia or even became flat. However, the number of adhered platelets on the NR-g-HEMA latex film decreased greatly with the grafting ratio up to 14%, and the platelets were disk shaped. The result indicated that the blood compatibility of NR latex film could be improved by grafting copolymerization with HEMA.
Conclusion
HEMA could be grafted copolymerization with NR latex using CHPO and TEPA as a redox initiator system. The appropriate amounts of crosslinking agent, initiator and monomer were 0.1phr, 0.2 phr and 20phr, respectively, and the optimum reaction conditions were 16ºC×8h. The water contact angle measurement and platelet adhesion evaluation indicated that the NR-g-HEMA latex had higher hydrophilicity and blood compatibility compared to the original natural rubber, which would extend the uses of NR in producing medical articles.
Acknowledgements
The authors wish to thank the financial support by the Natural Science Fund of Hainan Province (No.808200).
References
1. Coran, A. Y. - "Science and technology of rubber", Academic Press, London, chapt.7, p.291(1979).
2. March, J. - "Advanced organic chemistry", 2nd edn, McGraw-Hill International, London, chapt.15 (1977).
3. Streitwiesser, A. & Heathcock, C. H. - "Introduction to Organic Vhemistry", 2nd ed., Macmillan, New York, p.302 (1981).
4. Gelling, I. R. & Porter, M. - "Natural rubber science and technology", Oxford University press, Oxford, p.359 (1988).
5. Burfield, D. R.; Chew, L. C. & Gan, S. N. - Polymer, 17, p.713 (1976). http://dx.doi.org/10.1016/0032-3861(76)90214-7
6. Loadman Junior, R. M.; Saville, B.; Steer, M. & Tidd, B. K. - Chem. Commun, p.1167 (1972).
7. Hourston, D. J. & Romaine, J. - Eur. Polym. J., 25, p.695 (1989). http://dx.doi.org/10.1016/0014-3057(89)90031-1
8. Hourston, D. J. & Romaine, J. - J. Appl. Polym. Sci., 39, p.1587 (1990). http://dx.doi.org/10.1002/app.1990.070390715
9. Arayapranee, W.; Prasassarakich, P. & Rempel, G. L. - J. Appl. Polym. Sci., 89, p.63 (2003). http://dx.doi.org/10.1002/app.11999
10. George, B.; Maiti, S. N. & Varma, I. K. - J. Elastomers Plast., 38, p.319 (2006). http://dx.doi.org/10.1177/0095244306067129
11. Kochthongrasamee, T.; Prasassarakich, P. & Kiatkamjornwong, S. - J. Appl. Polym. Sci., 101, p.2587 (2006). http://dx.doi.org/10.1002/app.23997
12. Hourston, D. J. & Romaine, J. - J. Appl. Polym. Sci., 43, p.2207 (1991). http://dx.doi.org/10.1002/app.1991.070431208
13. Charmondusit, K.; Kiatkamjornwong, S. & Prasassarakich, P. - J. Sci. Chula. Univ., 23, p.167 (1998).
14. Kawahara, S.; Kawazura, T.; Sawada, T. & Isono, Y. - Polymer, 44, p.4527 (2003). http://dx.doi.org/10.1016/S0032-3861(03)00415-4
15. Chuayjuljit, S.; Siridamrong, P. & Pimpan, V. - J. Appl. Polym. Sci., 94, p.1496 (2004). http://dx.doi.org/10.1002/app.21064
16. Hu, R.; Dimonie, V. L. & El-Aasser, M. S. - J. Appl. Polym. Sci., 64, p.1123 (1997). http://dx.doi.org/10.1002/(SICI)1097-4628(19970509)64:6<1123::AID-APP12>3.0.CO;2-X
17. Okieimen, F. E. & Urhoghidei, I. N. - J. Appl. Polym. Sci., 84, p.1872 (2002). http://dx.doi.org/10.1002/app.10474
18. Fuminori, I.; Kimiko, M.; Hiroyuki, O.; Hiroshi, T. & Shinzo, O. - Colloids Surf. A: Physicochem. Eng. Aspects, 2, p.71 (2004).
19. Kangwansupamonkon, W.; Gilbert, R. G. & Kiatkamjornwong, S. - Macromo. Chem. Phys., 206, p.2450 (2005). http://dx.doi.org/10.1002/macp.200500255
20. Oliveira, P. C.; Guimarães, A.; Cavaillé, J-Y.; Chazeau, L. & Gilbert, R. G. - Polymer, 46, p.1105 (2005). http://dx.doi.org/10.1016/j.polymer.2004.11.048
21. Promdum, Y.; Klinpituksa, P. & Ruamcharoen, A. - Songklanakarin J. Sci. Technol., 31, p.453 (2009).
22. Li, J.; Geng, H. B. & Chen, Q. M. - Acta Polym. Sinica, 6, p.838 (2006). http://dx.doi.org/10.3724/SP.J.1105.2006.00838
23. Wang, P.; Tan, K. L.; Ho, C. C.; Khew, M. C. & Kang, E. T. - Eur. Polym. J., 36, p.1323 (2000). http://dx.doi.org/10.1016/S0014-3057(99)00193-7
24. Cheo, S. H. Y.; Wang, P.; Tan, K. L.; Ho, C. C. & Kang, E. T. - J. Mater. Sci.- Mater. Med., 12, p.377 (2001). http://dx.doi.org/10.1023/A:1011280416520
25. Kochthongrasamee, T.; Prasassarakich, P. & Kiatkamjornwong, S. - J. Appl. Polym. Sci., 101, p.2587 (2006). http://dx.doi.org/10.1002/app.23997
26. Anancharungsuk, W.; Tanpantree, S. & Sruangamurak, A. - J. Appl. Polym. Sci., 104, p.2270 (2007). http://dx.doi.org/10.1002/app.25661
27. Hoven, V. P., Chombanpaew, K.; Iwasaki, Y. & Tasakorn, P. - J. Appl. Polym. Sci., 112, p.208 (2009). http://dx.doi.org/10.1002/app.29408
28. Kwok, D. Y. & Neumann, A. W. - Adv. Colloid Interface Sci., 81, p.167 (1999). http://dx.doi.org/10.1016/S0001-8686(98)00087-6
Received: 10/20/13
Revised: 03/06/14
Accepted: 03/18/14
- 1. Coran, A. Y. - "Science and technology of rubber", Academic Press, London, chapt.7, p.291(1979).
- 2. March, J. - "Advanced organic chemistry", 2nd edn, McGraw-Hill International, London, chapt.15 (1977).
- 3. Streitwiesser, A. & Heathcock, C. H. - "Introduction to Organic Vhemistry", 2nd ed., Macmillan, New York, p.302 (1981).
- 4. Gelling, I. R. & Porter, M. - "Natural rubber science and technology", Oxford University press, Oxford, p.359 (1988).
- 5. Burfield, D. R.; Chew, L. C. & Gan, S. N. - Polymer, 17, p.713 (1976). http://dx.doi.org/10.1016/0032-3861(76)90214-7
- 6. Loadman Junior, R. M.; Saville, B.; Steer, M. & Tidd, B. K. - Chem. Commun, p.1167 (1972).
- 7. Hourston, D. J. & Romaine, J. - Eur. Polym. J., 25, p.695 (1989). http://dx.doi.org/10.1016/0014-3057(89)90031-1
- 8. Hourston, D. J. & Romaine, J. - J. Appl. Polym. Sci., 39, p.1587 (1990). http://dx.doi.org/10.1002/app.1990.070390715
- 9. Arayapranee, W.; Prasassarakich, P. & Rempel, G. L. - J. Appl. Polym. Sci., 89, p.63 (2003). http://dx.doi.org/10.1002/app.11999
- 10. George, B.; Maiti, S. N. & Varma, I. K. - J. Elastomers Plast., 38, p.319 (2006). http://dx.doi.org/10.1177/0095244306067129
- 11. Kochthongrasamee, T.; Prasassarakich, P. & Kiatkamjornwong, S. - J. Appl. Polym. Sci., 101, p.2587 (2006). http://dx.doi.org/10.1002/app.23997
- 12. Hourston, D. J. & Romaine, J. - J. Appl. Polym. Sci., 43, p.2207 (1991). http://dx.doi.org/10.1002/app.1991.070431208
- 13. Charmondusit, K.; Kiatkamjornwong, S. & Prasassarakich, P. - J. Sci. Chula. Univ., 23, p.167 (1998).
- 14. Kawahara, S.; Kawazura, T.; Sawada, T. & Isono, Y. - Polymer, 44, p.4527 (2003). http://dx.doi.org/10.1016/S0032-3861(03)00415-4
- 15. Chuayjuljit, S.; Siridamrong, P. & Pimpan, V. - J. Appl. Polym. Sci., 94, p.1496 (2004). http://dx.doi.org/10.1002/app.21064
- 17. Okieimen, F. E. & Urhoghidei, I. N. - J. Appl. Polym. Sci., 84, p.1872 (2002). http://dx.doi.org/10.1002/app.10474
- 18. Fuminori, I.; Kimiko, M.; Hiroyuki, O.; Hiroshi, T. & Shinzo, O. - Colloids Surf. A: Physicochem. Eng. Aspects, 2, p.71 (2004).
- 19. Kangwansupamonkon, W.; Gilbert, R. G. & Kiatkamjornwong, S. - Macromo. Chem. Phys., 206, p.2450 (2005). http://dx.doi.org/10.1002/macp.200500255
- 20. Oliveira, P. C.; Guimarães, A.; Cavaillé, J-Y.; Chazeau, L. & Gilbert, R. G. - Polymer, 46, p.1105 (2005). http://dx.doi.org/10.1016/j.polymer.2004.11.048
- 21. Promdum, Y.; Klinpituksa, P. & Ruamcharoen, A. - Songklanakarin J. Sci. Technol., 31, p.453 (2009).
- 22. Li, J.; Geng, H. B. & Chen, Q. M. - Acta Polym. Sinica, 6, p.838 (2006). http://dx.doi.org/10.3724/SP.J.1105.2006.00838
- 23. Wang, P.; Tan, K. L.; Ho, C. C.; Khew, M. C. & Kang, E. T. - Eur. Polym. J., 36, p.1323 (2000). http://dx.doi.org/10.1016/S0014-3057(99)00193-7
- 24. Cheo, S. H. Y.; Wang, P.; Tan, K. L.; Ho, C. C. & Kang, E. T. - J. Mater. Sci.- Mater. Med., 12, p.377 (2001). http://dx.doi.org/10.1023/A:1011280416520
- 25. Kochthongrasamee, T.; Prasassarakich, P. & Kiatkamjornwong, S. - J. Appl. Polym. Sci., 101, p.2587 (2006). http://dx.doi.org/10.1002/app.23997
- 26. Anancharungsuk, W.; Tanpantree, S. & Sruangamurak, A. - J. Appl. Polym. Sci., 104, p.2270 (2007). http://dx.doi.org/10.1002/app.25661
- 27. Hoven, V. P., Chombanpaew, K.; Iwasaki, Y. & Tasakorn, P. - J. Appl. Polym. Sci., 112, p.208 (2009). http://dx.doi.org/10.1002/app.29408
- 28. Kwok, D. Y. & Neumann, A. W. - Adv. Colloid Interface Sci., 81, p.167 (1999). http://dx.doi.org/10.1016/S0001-8686(98)00087-6
Publication Dates
-
Publication in this collection
10 June 2014 -
Date of issue
June 2014
History
-
Accepted
18 Mar 2014 -
Reviewed
06 Mar 2014 -
Received
20 Oct 2013