
Abstract
Short fibers are used in thermoplastic composites to increase their tensile and flexural resistance; however, it often decreases impact resistance. Composites with short vegetal fibers are not an exception to this behavior. The purpose of this work is to produce a vegetal fiber reinforced composite with improved tensile and impact resistance in relation to the polymer matrix. We used poly(ethylene-co-vinyl acetate), EVA, to recover the impact resistance of high density polyethylene, HDPE, reinforced with Curauá fibers, CF. Blends and composites were processed in a corotating twin screw extruder. The pure polymers, blends and composites were characterized by differential scanning calorimetry, thermogravimetry, infrared spectroscopy, scanning electron microscopy, tensile mechanical properties and Izod impact resistance. EVA used as impact modifier in the HDPE matrix exhibited a co-continuous phase and in the composites the fibers were homogeneously dispersed. The best combination of mechanical properties, tensile, flexural and impact, were obtained for the formulations of composites with 20 wt. % of CF and 20 to 40 wt. % of EVA. The composite prepared with 20 wt. % EVA and containing 30 wt. % of CF showed impact resistance comparable to pure HDPE and improved tensile and flexural mechanical properties.
Keywords:
fibers; impact behavior; mechanical testing; extrusion
1 Introduction
Vegetal fibers are largely replacing other reinforcing agents for thermoplastic and thermoset composites, because they are produced from renewable resources, have high toughness, have low density and are biodegradable[11 Netravali, A. N., & Chabba, S. (2003). Composites get greener. Materials Today, 6(4), 22-29. http://dx.doi.org/10.1016/S1369-7021(03)00427-9.
http://dx.doi.org/10.1016/S1369-7021(03)...
]. Their use produces composites with increased tensile and flexural mechanical properties in comparison to the matrix polymer. Additionally, thermoplastic composites with vegetal fibers cause less wear to the processing equipment in comparison to glass fibers[22 Bledzki, A. K., & Gassan, J. (1999). Composites reinforced with cellulose based fibers. Progress in Polymer Science, 24(2), 221-274. http://dx.doi.org/10.1016/S0079-6700(98)00018-5.
http://dx.doi.org/10.1016/S0079-6700(98)...
,33 Pandey, J. K., Ahn, S. H., Lee, C. S., Mohanty, A. K., & Misra, M. (2010). Recent advances in the application of natural fiber based composites. Macromolecular Materials and Engineering, 295(11), 975-989. http://dx.doi.org/10.1002/mame.201000095.
http://dx.doi.org/10.1002/mame.201000095...
].
The vegetal fibers used in this work, Curauá fibers, CF, extracted from the leaves of an Amazonian plant of the bromeliad family (Ananas erectfolius L. B. Smith) are produced on a large scale. Like other lignocellulosic materials, are composed of hemicellulose, cellulose and lignin. We and other authors, previously characterized these fibers for their mechanical properties and chemical composition[44 Tomczak, F., Satyanarayana, K. G., & Sydenstricker, T. H. D. (2007). Studies on lignocellulosic fibers of Brazil: Part III – Morphology and properties of Brazilian curauá fibers. Composites. Part A, Applied Science and Manufacturing, 38(10), 2227-2236. http://dx.doi.org/10.1016/j.compositesa.2007.06.005.
http://dx.doi.org/10.1016/j.compositesa....
,55 Spinacé, M. A. S., Lambert, C. S., Fermoselli, K. K. G., & De Paoli, M. A. (2009). Characterization of lignocellulosic Curaua fibres. Carbohydrate Polymers, 77(1), 47-53. http://dx.doi.org/10.1016/j.carbpol.2008.12.005.
http://dx.doi.org/10.1016/j.carbpol.2008...
]. Their specific tensile mechanical properties are similar to those of glass fibers, making them a good candidate for their substitution[22 Bledzki, A. K., & Gassan, J. (1999). Composites reinforced with cellulose based fibers. Progress in Polymer Science, 24(2), 221-274. http://dx.doi.org/10.1016/S0079-6700(98)00018-5.
http://dx.doi.org/10.1016/S0079-6700(98)...
]. The diameter of the pristine CF is in the range of 30 to 100 μm and each fiber is composed of a bundle of microfibers, which are fibrillated upon extrusion, as demonstrated in a previous work[66 Araujo, J. R., Mano, B., Teixeira, G. M., Spinacé, M. A. S., & De Paoli, M. A. (2010). Biomicrofibrilar composites of high density polyethylene reinforced with Curauá fibers: Mechanical, interfacial and morphological properties. Composites Science and Technology, 70(11), 1637-1644. http://dx.doi.org/10.1016/j.compscitech.2010.06.006.
http://dx.doi.org/10.1016/j.compscitech....
]. The extent of fibrillation and fiber breakage is strongly dependent on the processing conditions[77 Mano, B., Araujo, J. R., Spinacé, M. A. S., & De Paoli, M. A. (2010). Polyolefin composites with curaua fibres: Effect of the processing conditions on mechanical properties, morphology and fibres dimensions. Composites Science and Technology, 70(1), 29-35. http://dx.doi.org/10.1016/j.compscitech.2009.09.002.
http://dx.doi.org/10.1016/j.compscitech....
].
Cellulose acetate[88 Gutiérrez, M. C., De Paoli, M. A., & Felisberti, M. I. (2012). Biocomposites based on cellulose acetate and short Curauá fibers: Effect of plasticizers and chemical treatments of the fibers. Composites. Part A, Applied Science and Manufacturing, 43(8), 1338-1346. http://dx.doi.org/10.1016/j.compositesa.2012.03.006.
http://dx.doi.org/10.1016/j.compositesa....
], polypropylene[99 Mano, B., Araujo, J. R., Waldman, W. R., Spinacé, M. A. S., & De Paoli, M. A. (2013). Mechanical properties, morphology and thermal degradation of a biocomposite of polypropylene and Curauá fibers: coupling agent effect. Polímeros: Ciência e Tecnologia, 23, 161-168. http://dx.doi.org/10.4322/S0104-14282013005000025.
http://dx.doi.org/10.4322/S0104-14282013...
], post-consumer polypropylene[1010 Spinace, M. A. S., Fermoselli, K. K. G., & De Paoli, M. A. (2009). Recycled polypropylene reinforced with Curauá fibers by extrusion. Journal of Applied Polymer Science, 112(6), 3686-3694. http://dx.doi.org/10.1002/app.29683.
http://dx.doi.org/10.1002/app.29683...
], polyamide-6[1111 Santos, P. A., Spinacé, M. A. S., Fermoselli, K. K. G., & De Paoli, M. A. (2009). Efeito da forma de processamento e do tratamento da fibra de curauá nas propriedades de compósitos com poliamida-6. Polímeros: Ciência e Tecnologia, 19(1), 31-39. http://dx.doi.org/10.1590/S0104-14282009000100010.
http://dx.doi.org/10.1590/S0104-14282009...
] and high-density polyethylene[66 Araujo, J. R., Mano, B., Teixeira, G. M., Spinacé, M. A. S., & De Paoli, M. A. (2010). Biomicrofibrilar composites of high density polyethylene reinforced with Curauá fibers: Mechanical, interfacial and morphological properties. Composites Science and Technology, 70(11), 1637-1644. http://dx.doi.org/10.1016/j.compscitech.2010.06.006.
http://dx.doi.org/10.1016/j.compscitech....
] were reinforced with CF in our group by processing in a corotating twin-screw extruder. In all cases, the tensile and flexural properties were improved and 20 wt. % of CF was the best content in the composites to achieve this improvement. In most cases, the use of a coupling agent provided a better fiber to matrix interaction. Like in other polymer/fiber composites, tensile and flexural mechanical properties improvement is a consequence of stress transference to the fiber, provided by a good fiber to matrix adhesion. However, this improvement occurs in parallel to a decrease in the impact resistance, as generally observed for short fiber reinforced thermoplastic polymers[1212 Bader, M. G. (1994). Short fibre reinforced thermoplastics. In F. R. Jones. Handbook of polymer-fibre composites (pp. 275-277). Essex: Longman Scientific and Technical. http://dx.doi.org/10.1002/pat.1995.220060807.
http://dx.doi.org/10.1002/pat.1995.22006...
].
High-density polyethylene, HDPE, has a high impact resistance, however, when reinforced with vegetal fibers this resistance decreases by ca. 30%[1313 Spinace, M. A. S., Janeiro, L. G., Bernardino, F. C., Grossi, T. A., & De Paoli, M. A. (2011). Polyolefins reinforced with short vegetal fibers: Sisal vs. Curauá. Polímeros: Ciência e Tecnologia, 21, 168-174. http://dx.doi.org/10.1590/S0104-14282011005000036.
http://dx.doi.org/10.1590/S0104-14282011...
]. The decrease in impact resistance was also observed for composites of high-density biopolyethylene with Curauá fibers, in different proportions and processed by two different methods[1414 Castro, D. O., Ruvolo-Filho, A., & Frollini, E. (2012). Materials prepared from biopolyethylene and curaua fibers: composites from biomass. Polymer Testing, 31(7), 880-888. http://dx.doi.org/10.1016/j.polymertesting.2012.05.011.
http://dx.doi.org/10.1016/j.polymertesti...
]. To attain the impact resistance of the neat biopolyethylene the same authors used liquid hydroxylated polybutadiene, as compatibilizer and impact modifier, in the composite with Curauá fibers. Similar impact resistance reduction and use of impact modifiers was reported for short jute fiber reinforced polypropylene composites[1515 Rana, A. K., Mandal, A., & Bandyopadhyay, S. (2003). Bandyopadhyay S.: Short jute fiber reinforced polypropylene composites: effect of compatibilizer, impact modifier and fiber loading. Composites Science and Technology, 63(6), 801-806. http://dx.doi.org/10.1016/S0266-3538(02)00267-1.
http://dx.doi.org/10.1016/S0266-3538(02)...
].
The most common strategy to improve the impact resistance, i.e. toughness, of a thermoplastic material is to make a blend with an elastomer. This is routinely used on an industrial scale, like when polycarbonate becomes tougher by blending with high impact polystyrene (Noryl™ is the commercial name of this blend). To obtain the tougher material it is necessary to make an immiscible and compatible blend, controlling the concentration of the modifier, particles size, distance between the particles and degree of adhesion between the polymeric phases[1616 Rabello, M., & De Paoli, M.-A. (2013). Aditivação de termoplásticos. São Paulo: Artliber.]. In this case, the rubbery phase concentrates or absorbs tension, changing the tension states of the matrix. The tension absorbed by the modifier causes plastic deformation, which absorbs the impact.
Previous works report on the rheology, morphology and mechanical properties of polyethylene blended with poly(ethylene-co-vinyl acetate) in different compositions and using various comonomer contents[1717 Faker, M., Razavi Aghjeh, M. K., Ghaffari, M., & Seyyedi, S. A. (2008). Rheology, morphology and mechanical properties of polyethylene/ethylene vinyl acetate copolymer (PE/EVA) blends. European Polymer Journal, 44(6), 1834-1842. http://dx.doi.org/10.1016/j.eurpolymj.2008.04.002.
http://dx.doi.org/10.1016/j.eurpolymj.20...
18 Khonakdar, H. A., Wagenknecht, U., Jafari, S. H., Hässler, R., & Eslami, H. (2004). Dynamic mechanical properties and morphology of polyethylene/ethylene vinyl acetate copolymer blends. Advances in Polymer Technology, 23(4), 307-315. http://dx.doi.org/10.1002/adv.20019.
http://dx.doi.org/10.1002/adv.20019...
-1919 Khonakdar, H. A., Jafari, S. H., Yavari, A., Asadinezhad, A., & Wagenknecht, U. (2005). Rheology, Morphology and estimation of interfacial tension of LDPE/EVA and HDPE/EVA blends. Polymer Bulletin, 54(1-2), 75-84. http://dx.doi.org/10.1007/s00289-005-0365-6.
http://dx.doi.org/10.1007/s00289-005-036...
]. These works focused on the miscibility of the blends but did not report on the effect of EVA content on the impact resistance of the blend.
As discussed above, vegetal fiber composites of HDPE have improved tensile and flexural mechanical properties in comparison to pure HDPE; however, the drawback is the reduction of the impact resistance. Thus, in the area of thermoplastic biocomposites the existing gap is a material that combines high tensile and flexural resistance with good impact resistance. Making the composite with a blend of HDPE with an immiscible softer material can recuperate this property. Thus, the aim of the present work is to produce a composite material associating the high tensile and flexural strength of the composite of HDPE with CF and the good impact resistance of pure HDPE. This is pursued by using EVA as impact modifier for the composite of HDPE with CF.
2 Materials and Methods
2.1 Material used
CF supplied by Embrapa-PA (Belém, Brazil) were fractioned in a three-knife rotary mill (Rone, model NFA 1533), with knife/counter-knife gap of 0.5 mm, washed two times with tap water, dried in open air for 24 h and further dried in a conventional oven at 100 °C for 8 h. Before processing, the fibers were disaggregated in a mechanical homogenizer (MH Equipamentos) for 2 min, to facilitate the feeding into the side-feeder of the extruder.
HDPE, JV060U, was supplied by Braskem (Triunfo, Brazil) with melt flow index of 6.1 - 8.0 g 10 min-1 at 190 °C and 2.16 kg and ρ = 0.957 g cm-3. EVA, HM2528, was from Braskem (Triunfo, Brazil) with 28 wt. % of vinyl acetate and melt flow index of 25 g 10 min-1 at 190 °C and 2.16 kg.
2.2 Processing
Blends and composites were processed in a corotating intermeshing twin-screw extruder (Coperion Werner & Pfleiderer, Germany, ZSK 26 Mc, L/D = 44) with degassing and a side-feeder. Feeding was controlled with two Brabender DDW-MD2-DSR28-10 gravimetric dosimeters. For composites processing we used a previously published screw design[2020 Araujo, J. R., Waldman, W. R., & De Paoli, M. A. (2008). Thermal properties of high density polyethylene composites with natural fibres: Coupling agent effect. Polymer Degradation & Stability, 93(10), 1770-1775. http://dx.doi.org/10.1016/j.polymdegradstab.2008.07.021.
http://dx.doi.org/10.1016/j.polymdegrads...
], while, for the blends, part of the kneading elements were replaced by transport elements to reduce shear. The composition of the blends and composites is shown in weight percent, wt. %, in Table 1.
Composition of blends and composites in wt. %. HDPE = high density polyethylene, EVA = poly(ethylene-co-vinyl acetate) and CF = Curauá fiber.
Blends were prepared by pre-mixing 20, 30 or 40 wt. % of EVA into HDPE and extrusion in the following conditions: temperature profile of 170, 175, 180, 185, 190, 190, 195, 200, 200, 195 and 200 ºC, from feed to die, and screw rotation of 300 rpm. The average torque and mass temperature were 30% and 208 °C, respectively.
Composites of all the HDPE/EVA blends were processed by extrusion with 20 or 30 wt. % of fibers using a temperature profile of 120, 120,125, 125, 130, 130, 135, 135, 140, 135 and 130 °C, from feed to die, and screw rotation of 300 rpm. Fibers were fed using the side-feeder, at 265 rpm, and vacuum degassing was employed in all processes. The average torque and mass temperature were 47% and 151 °C, respectively.
Test samples with dimensions according to ASTM D-638 and ASTM D-256 were prepared by injection-molding (Arburg, All-rounder M250) using previously dried (100 ºC for 1 h) pellets of the blends and composites. For the blends the conditions were: temperature profile of 180, 185, 190, 200 and 205 °C, 1.4 x 108 Pa of injection pressure, 1.0 x 108 Pa of hold pressure, injection velocity of 15 cm3 s-1, mold temperature of 20 °C and 20 s of cooling time. For the composites with 20 wt. % of fibers the conditions were: temperature profile of 150,160, 170, 175 and 165 °C, 1.2 x 108 Pa of injection pressure, 8.0 x 107 Pa of hold pressure, injection velocity of 15 cm3 s-1, mold temperature of 20 °C and 10 s of cooling time. For composites with 30 wt. % of fibers the conditions were: temperature profile of 170, 180, 190, 200 and 210 °C, 1.5 x 108 Pa of injection pressure, 1.3 and 1.4 x 107 Pa of hold pressure, injection velocity of 3 cm3 s-1, mold temperature of 20 °C and 10 s of cooling time.
2.3 Characterization of blends and composites
Differential scanning calorimetry, DSC, was used to determine the melting temperature, Tm, and glass transition temperature, Tg, using a DSC-Q100 equipment (TA Instruments) with a cooling and heating rate of 10 °C cm-1 in the temperature range of -50 to 200 °C, under an argon atmosphere (50 mL min-1).
Thermogravimetric analyses, TGA, was used to determine the mass loss parameters using a TA2900 (TA Instruments) in the temperature range from 20 to 600 °C, with a heating rate of 10 °C cm-1 under synthetic air flow (100 mL min-1).
Scanning electron microscopy, SEM, was used to monitor the morphology of the cryogenic fracture surface of the samples. For this, the injection molded test samples were maintained for 30 min in liquid N2 before fracturing. The surface of the fractures were coated with carbon and gold by sputtering using a Balzers MD BalTec 020 equipment. SEM analyses were done at an accelerating voltage of 5 kV using a Jeol JSM–6360LV equipment.
Fourier transform infrared, FTIR, spectra in ATR mode were obtained using a Smiths IlluminatIR II-Micro-Infrared Probe for Optical Microscopy, range 4000 - 400 cm-1, 21 scans min-1 and resolution of 4 cm-1. The spectra of pure HDPE and blends with EVA were obtained by ATR using samples in the form of films, produced by pressing in a hydraulic press (Marconi MA-098-A) at 160 °C and 0.02 MPa for 2 min., For the composites, the samples were milled to observe the contribution of the fibers to the spectra measured as KBr pellets in the transmittance mode.
For mechanical properties determinations, the samples were conditioned for 48 h in an acclimatized room at 25 (± 5) °C and 50 (± 5) % relative humidity. The tensile properties were determined according to ASTM D-638 using an EMIC DL2000 equipment with a 5 kN load cell. For the impact resistance we used the ASTM D-256 standard and an EMIC AlC1 equipment with a 2.7 J hammer.
3 Results and Discussions
3.1 HDPE/EVA Blend
3.1.1 Differential Scanning Calorimetry
From the DSC curves of the blends in the second heating scan, Figure 1a, we determined the melting temperatures, Tm, as the peak of the endothermic transition. The Tm of the HDPE phase in the blends did not change with increased contents of EVA, in relation to pure HDPE. The first heating is routinely used in DSC measurements to erase the thermal history of the sample. These curves also show that there was no shift of the glass transition temperature (Tg) of the EVA phases in the blends, as highlighted in Figure 1b for the HDPE/40EVA blend. The Tg of the HDPE phase was not detected in the blends because it is below the temperature range used (ca. - 95 °C). These DSC results demonstrate that, according to the Tg criteria, EVA and HDPE form immiscible phases in these blends. By determining the area under the melting peak, it is concluded that the presence of EVA does not change the degree of crystallinity of the HDPE phase.

a) DSC curves (2nd heating) of HDPE blends with 20, 30 and 40 wt. % of EVA and (b) detail of the part of the curve used for Tg determination in a) for HDPE/40EVA. Curves in a) were vertically shifted for better comparison.
3.1.2 Thermogravimetry
In Figure 2, we compare the thermogravimetric curves of HDPE, EVA and the blend HDPE/20EVA, measured under synthetic air flow. Other blend compositions showed similar behaviors. The onset mass loss in these curves (ca. 250 oC) corresponds to the oxidative thermal degradation onset of pure HDPE, the polyethylene block of EVA and the HDPE phase of the blend. The inflections observed in the pure HDPE curve are a consequence of the formulation of the grade used. The TGA curve of EVA reaches a plateau at ca. 70% mass, with a subsequent sharp drop to 15% at 440 oC, corresponding to the vinyl acetate block oxidative thermal degradation with the formation of acetic acid and conjugated double bonds[2121 McNeill, I. C. (1997). Thermal degradation mechanisms of some addition polymers and copolymers. Journal of Analytical and Applied Pyrolysis, 40-41, 21-41. http://dx.doi.org/10.1016/S0165-2370(97)00006-5.
http://dx.doi.org/10.1016/S0165-2370(97)...
]. Residue formation above 450 oC is similar for all three samples. The blend curve suggests that there is no interaction between the thermo oxidation reactions of the blend components.
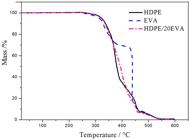
Comparison of the thermogravimetric curves of HDPE, EVA and the blend HDPE/20EVA, measured under synthetic air flow at 10 oC min-1.
3.1.3 Infrared spectroscopy
Figure 3 illustrates the reflectance FTIR spectra of HDPE, EVA and the blend HDPE/20EVA (all blend compositions were measured) measured directly at the surface of the injection-molded specimens. The infrared spectrum provides information about structural aspects of polymers, such as chemical composition, conformation and structural configuration. The position and relative intensities of the absorption bands may give information concerning the interaction between the blend components. We observe that the reflectance infrared spectra of the blends correspond exactly to a superimposition of the spectra of its components with no band shifts. This is another indication for the lack of chemical interaction between the blend components, corroborating the conclusion that it is an immiscible blend.

ATR-FTIR spectra for EVA, HDPE, and the blend of HDPE/20EVA. Spectra vertically shifted for comparison.
3.1.4 Scanning Electron Microscopy
Figure 4 shows the SEM micrographs of the cryofractured surface of injection molded samples of the HDPE/EVA blends with 30 and 40 wt. % of EVA. Figure 4a shows the micrograph of the crude fracture and in Figure 4b the EVA phase was extracted with acetone to highlight the morphology. As shown in the micrographs, the HDPE/EVA blends exhibit a co-continuous morphology, with EVA as dispersed phase and HDPE as continuous phase. This phase separation confirms the immiscibility of the blend components. SEM micrographs of the other blends were also obtained and show the same features. The extraction of EVA with acetone resulted in “empty” spaces homogenously distributed in the HDPE matrix, Figure 4b, confirming that EVA is uniformly distributed in the HDPE phase. The transmittance FTIR spectrum of a film cast from the acetone extract corresponds to the spectrum of EVA, confirming its extraction.

SEM micrographs of the fracture surface of injection molded blends: (a) HDPE/30 EVA with scale bar of 1 µm and (b) HDPE/40EVA after extraction with acetone with scale bar of 10 µm.
3.1.5 Mechanical properties
We measured the tensile properties of the blends to determine the effect of the EVA concentration on the tenacity. Figure 5 shows the average stress-strain curves of pure HDPE and the blends. Figure 6 compares the tensile strength and modulus for the blends and pure HDPE (values shown in Table 3). The approximately linear decrease of tensile strength and modulus with EVA concentration in the blends also reflects their immiscibility.

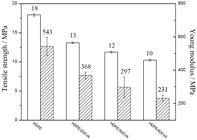
Comparison of tensile strength
 and Young modulus
and Young modulus  for the HDPE/EVA blends, as a function of EVA content.
for the HDPE/EVA blends, as a function of EVA content.
Tensile and impact resistance results for pure HDPE, HDPE/EVA blends and blends reinforced with Curauá fiber. (* from ref.[66 Araujo, J. R., Mano, B., Teixeira, G. M., Spinacé, M. A. S., & De Paoli, M. A. (2010). Biomicrofibrilar composites of high density polyethylene reinforced with Curauá fibers: Mechanical, interfacial and morphological properties. Composites Science and Technology, 70(11), 1637-1644. http://dx.doi.org/10.1016/j.compscitech.2010.06.006.
http://dx.doi.org/10.1016/j.compscitech.... ]).
The tenacity in Table 2 was calculated as the area under the stress-strain curves. Comparing to pure HDPE, there is a large increase in tenacity in the HDPE/20EVA blend. This value slightly decreases for HDPE/30EVA and significantly drops when the concentration of EVA is increased in the HDPE/40EVA blend. This indicates that 30 wt. % of EVA is the upper limit of concentration to increase tenacity of HDPE with an EVA copolymer containing 28% of vinyl acetate.
Tenacity, calculated as the integrated area under the stress-strain curves for HDPE and the blends.
3.2 HDPE/EVA blends reinforced with Curauá fibers
3.2.1 SEM
The SEM micrographs of the cryogenic fracture of the injection molded samples of the composites of the HDPE blend with 20, 30 and 40 wt. % EVA and 20 wt. % CF are depicted in Figure 7. In Figure 7a we observe that the incorporation of the fibers in the blend did not change its morphology, with a co-continuous phase similar to the pure blend, shown in Figure 4a. Figure 7b illustrates the homogeneous distribution of the fibers in the blend and their adhesion to the matrix, evidenced by low occurrence of fiber pullout during fracture. The good fiber-matrix adhesion shown in Figs 7a and highlighted by a circle, confirms the good interaction between the blend and the reinforcing agent. Higher magnification (not shown) reveals that the fibers are partially covered by the polymer. The micrograph of the cryogenically fractured composite extracted with acetone is shown in Figure 7d with higher magnification. Some voids observed between the fibers and the matrix in Figure 7d can be an indication that the EVA phase is responsible for the good fiber/matrix adhesion in the blend. This argument is reinforced by the absence of holes left by EVA extraction in Figure 7d, when compared to Figure 4b.

SEM micrographs of the cryogenic fracture surface of injection molded composites: (a) HDPE/20EVA20CF showing fiber-matrix interaction and co-continuous EVA phase, (b) HDPE/30EVA20CF showing the homogeneous distribution of the fibers, (c) HDPE/40EVA20CF highlighting fiber diameter and (d) HDPE/40EVA20CF composite after extraction with acetone with higher magnification.
The diameter of the pristine Curauá fibers is ca. 100 µm and it is reduced by fiber fibrillation, occuring as a consequence of the high shear of the twin-screw extruder, as reported in previous works[66 Araujo, J. R., Mano, B., Teixeira, G. M., Spinacé, M. A. S., & De Paoli, M. A. (2010). Biomicrofibrilar composites of high density polyethylene reinforced with Curauá fibers: Mechanical, interfacial and morphological properties. Composites Science and Technology, 70(11), 1637-1644. http://dx.doi.org/10.1016/j.compscitech.2010.06.006.
http://dx.doi.org/10.1016/j.compscitech....
]. The presence of microfibrils in the SEM micrographs, with diameters of the order of 5 µm or less, observed in Figures 7c and 7d, confirm that the CF fibers were fibrillated during processing. This diameter reduction reflects in a high aspect ratio of the fibers, improving the reinforcement effect[2222 Ausias, G., Bourmaud, A., Coroller, G., & Baley, C. (2013). Study of the fibre morphology stability in polypropylene-flax composites. Polymer Degradation & Stability, 98(6), 1216-1224. http://dx.doi.org/10.1016/j.polymdegradstab.2013.03.006.
http://dx.doi.org/10.1016/j.polymdegrads...
].
3.2.2 Infrared spectrophotometry
Figure 8 shows the transmittance infrared spectra of the composites of the HDPE/EVA blend with 20, 30 and 40 wt. % of EVA and 20 wt. % of Curauá fibers. These spectra were measured after dispersing the milled composites in KBr. This was done because the reflectance spectra of the surface would show no evidence for the presence of the fibers, which tend to migrate to the bulk of the material during injection molding[1212 Bader, M. G. (1994). Short fibre reinforced thermoplastics. In F. R. Jones. Handbook of polymer-fibre composites (pp. 275-277). Essex: Longman Scientific and Technical. http://dx.doi.org/10.1002/pat.1995.220060807.
http://dx.doi.org/10.1002/pat.1995.22006...
]. Bands labeled 1 to 8 correspond to HDPE and EVA in the blend, particularly band 6 at 1746 cm-1, corresponding to the C=O stretching of the acetate group of the EVA phase and band 1 at 727 cm-1, corresponding to the CH2 bending of HDPE and EVA. The presence of the fibers in the composites is evidenced by the bands labeled as a, b and c. The most evident is the broad band centered at 3440 cm-1, band c, corresponding to the O-H stretching of the cellulose and lignin hydroxyl groups, present in the fibers[44 Tomczak, F., Satyanarayana, K. G., & Sydenstricker, T. H. D. (2007). Studies on lignocellulosic fibers of Brazil: Part III – Morphology and properties of Brazilian curauá fibers. Composites. Part A, Applied Science and Manufacturing, 38(10), 2227-2236. http://dx.doi.org/10.1016/j.compositesa.2007.06.005.
http://dx.doi.org/10.1016/j.compositesa....
].

Transmittance infrared spectra of the composites of HDPE/EVA blends, with different EVA contents, and 20 wt. % of CF, measured as KBr pellets. Spectra were vertically shifted for comparison.
3.2.3 Thermogravimetry
Figure 9 shows the TGA curve for the composite of the HDPE blend with 20 wt. % of EVA and 20 wt. % of CF, HDPE/20EVA20CF. The curve shows two main degradation processes starting at ca. 250 and 400 °C. The first is assigned to thermal decomposition of the fiber, by comparing to other previously prepared composites[66 Araujo, J. R., Mano, B., Teixeira, G. M., Spinacé, M. A. S., & De Paoli, M. A. (2010). Biomicrofibrilar composites of high density polyethylene reinforced with Curauá fibers: Mechanical, interfacial and morphological properties. Composites Science and Technology, 70(11), 1637-1644. http://dx.doi.org/10.1016/j.compscitech.2010.06.006.
http://dx.doi.org/10.1016/j.compscitech....
], and the second to the blend, as compared to Figure 2. Similar to previous results reported in the literature[2323 Albano, C., Gonzalez, J., Ichazo, M., & Kaiser, D. (1999). Thermal stability of blends of polyolefins and sisal fiber. Polymer Degradation & Stability, 66(2), 179-190. http://dx.doi.org/10.1016/S0141-3910(99)00064-6.
http://dx.doi.org/10.1016/S0141-3910(99)...
], good fiber to matrix interaction favors the stability of the composite, shifting the matrix onset degradation temperature to a higher value. The absence of residue above 520 oC indicates absence of mineral contaminants in the fibers. The first derivative curve (not shown) indicates no interaction between the degradation products of the fibers and the blend. The TGA curves of the remaining composites show similar features.
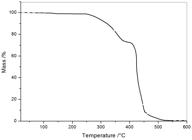
3.2.4 Tensile properties
Figure 10 and Table 3 present the variation of the mechanical properties of the composites with 20 and 30 wt. % of CF and toughened with different concentrations of EVA, in comparison to pure HDPE, to its composite with 20 wt. % of CF[66 Araujo, J. R., Mano, B., Teixeira, G. M., Spinacé, M. A. S., & De Paoli, M. A. (2010). Biomicrofibrilar composites of high density polyethylene reinforced with Curauá fibers: Mechanical, interfacial and morphological properties. Composites Science and Technology, 70(11), 1637-1644. http://dx.doi.org/10.1016/j.compscitech.2010.06.006.
http://dx.doi.org/10.1016/j.compscitech....
] and blends with different EVA contents. As observed previously, there is a strong increase of tensile strength and modulus by dispersing 20 wt. % of fibers in HDPE. When making the composites with the blends and 20 wt. % of fibers, these values decrease with the increased concentration of EVA in the blend and are recovered by increasing fiber content to 30 wt. %. Thus, the composite with 50 wt. % of HDPE, 20 wt. % of EVA and 30 wt. % of CF (HDPE/20EVA/30CF) shows the same tensile strength and a higher modulus in comparison to HDPE/20EVA/20CF, which contains 60 wt. % of HDPE. Thus, increasing the fiber content compensates for the effect of EVA on the tenacity of the matrix.

Comparison of the mechanical properties of pure HDPE, HDPE/20CF composite and composites of blends with different EVA and CF contents in wt. %: a) tensile strength
and Young’s modulus and b) impact resistance.
3.2.5 Impact resistance
Figure 10b and Table 3 show the variation of the impact resistance of the composites with 20 wt. % of CF and 20, 30 or 40 wt. % of EVA and for the composite with 30 wt. % of CF and 20 wt. % of EVA (HDPE/20EVA/30CF). It is clear that all composites with the HDPE/EVA blend have a higher impact resistance in comparison to pure HDPE. This is due to the EVA phase of the blend, which is responsible for the impact force dissipation. Additionally, increasing the fiber content in the 20EVA composite to 30 wt. % (HDPE/20EVA/30CF) yields a material with impact resistance similar to pure HDPE. Note that in this composite the HDPE content is only 50 wt. %.
4 Conclusions
HDPE and EVA form an immiscible blend with a high impact resistance in comparison to pure HDPE, because the dispersed EVA phase absorbs the impact energy. By using this blend to make composites with Curauá fibers, it is possible to obtain a material with good tensile resistance and, simultaneously, an impact resistance comparable to that of pure HDPE. This demonstrates that the mechanical properties of vegetal fibers based thermoplastic composites can be tailored for high impact resistant material applications. It is also important to note that the composite with the best mechanical properties uses 70% of petrochemical based raw materials and 30 wt. % of a renewable resource, with clear environmental benefits.
5. Acknowledgements
The authors thank EMBRAPA-PA for the fibers, Braskem for the polymer samples and FAPESP for financial support (Proc. 2010/17804-7). JAM thank SAE-Unicamp for a scholarship. We also thank Profs. Maria do Carmo Gonçalves and Maria Isabel Felisberti for suggestions.
6. References
-
1Netravali, A. N., & Chabba, S. (2003). Composites get greener. Materials Today, 6(4), 22-29. http://dx.doi.org/10.1016/S1369-7021(03)00427-9
» http://dx.doi.org/10.1016/S1369-7021(03)00427-9 -
2Bledzki, A. K., & Gassan, J. (1999). Composites reinforced with cellulose based fibers. Progress in Polymer Science, 24(2), 221-274. http://dx.doi.org/10.1016/S0079-6700(98)00018-5
» http://dx.doi.org/10.1016/S0079-6700(98)00018-5 -
3Pandey, J. K., Ahn, S. H., Lee, C. S., Mohanty, A. K., & Misra, M. (2010). Recent advances in the application of natural fiber based composites. Macromolecular Materials and Engineering, 295(11), 975-989. http://dx.doi.org/10.1002/mame.201000095
» http://dx.doi.org/10.1002/mame.201000095 -
4Tomczak, F., Satyanarayana, K. G., & Sydenstricker, T. H. D. (2007). Studies on lignocellulosic fibers of Brazil: Part III – Morphology and properties of Brazilian curauá fibers. Composites. Part A, Applied Science and Manufacturing, 38(10), 2227-2236. http://dx.doi.org/10.1016/j.compositesa.2007.06.005
» http://dx.doi.org/10.1016/j.compositesa.2007.06.005 -
5Spinacé, M. A. S., Lambert, C. S., Fermoselli, K. K. G., & De Paoli, M. A. (2009). Characterization of lignocellulosic Curaua fibres. Carbohydrate Polymers, 77(1), 47-53. http://dx.doi.org/10.1016/j.carbpol.2008.12.005
» http://dx.doi.org/10.1016/j.carbpol.2008.12.005 -
6Araujo, J. R., Mano, B., Teixeira, G. M., Spinacé, M. A. S., & De Paoli, M. A. (2010). Biomicrofibrilar composites of high density polyethylene reinforced with Curauá fibers: Mechanical, interfacial and morphological properties. Composites Science and Technology, 70(11), 1637-1644. http://dx.doi.org/10.1016/j.compscitech.2010.06.006
» http://dx.doi.org/10.1016/j.compscitech.2010.06.006 -
7Mano, B., Araujo, J. R., Spinacé, M. A. S., & De Paoli, M. A. (2010). Polyolefin composites with curaua fibres: Effect of the processing conditions on mechanical properties, morphology and fibres dimensions. Composites Science and Technology, 70(1), 29-35. http://dx.doi.org/10.1016/j.compscitech.2009.09.002
» http://dx.doi.org/10.1016/j.compscitech.2009.09.002 -
8Gutiérrez, M. C., De Paoli, M. A., & Felisberti, M. I. (2012). Biocomposites based on cellulose acetate and short Curauá fibers: Effect of plasticizers and chemical treatments of the fibers. Composites. Part A, Applied Science and Manufacturing, 43(8), 1338-1346. http://dx.doi.org/10.1016/j.compositesa.2012.03.006
» http://dx.doi.org/10.1016/j.compositesa.2012.03.006 -
9Mano, B., Araujo, J. R., Waldman, W. R., Spinacé, M. A. S., & De Paoli, M. A. (2013). Mechanical properties, morphology and thermal degradation of a biocomposite of polypropylene and Curauá fibers: coupling agent effect. Polímeros: Ciência e Tecnologia, 23, 161-168. http://dx.doi.org/10.4322/S0104-14282013005000025
» http://dx.doi.org/10.4322/S0104-14282013005000025 -
10Spinace, M. A. S., Fermoselli, K. K. G., & De Paoli, M. A. (2009). Recycled polypropylene reinforced with Curauá fibers by extrusion. Journal of Applied Polymer Science, 112(6), 3686-3694. http://dx.doi.org/10.1002/app.29683
» http://dx.doi.org/10.1002/app.29683 -
11Santos, P. A., Spinacé, M. A. S., Fermoselli, K. K. G., & De Paoli, M. A. (2009). Efeito da forma de processamento e do tratamento da fibra de curauá nas propriedades de compósitos com poliamida-6. Polímeros: Ciência e Tecnologia, 19(1), 31-39. http://dx.doi.org/10.1590/S0104-14282009000100010
» http://dx.doi.org/10.1590/S0104-14282009000100010 -
12Bader, M. G. (1994). Short fibre reinforced thermoplastics. In F. R. Jones. Handbook of polymer-fibre composites (pp. 275-277). Essex: Longman Scientific and Technical. http://dx.doi.org/10.1002/pat.1995.220060807
» http://dx.doi.org/10.1002/pat.1995.220060807 -
13Spinace, M. A. S., Janeiro, L. G., Bernardino, F. C., Grossi, T. A., & De Paoli, M. A. (2011). Polyolefins reinforced with short vegetal fibers: Sisal vs. Curauá. Polímeros: Ciência e Tecnologia, 21, 168-174. http://dx.doi.org/10.1590/S0104-14282011005000036
» http://dx.doi.org/10.1590/S0104-14282011005000036 -
14Castro, D. O., Ruvolo-Filho, A., & Frollini, E. (2012). Materials prepared from biopolyethylene and curaua fibers: composites from biomass. Polymer Testing, 31(7), 880-888. http://dx.doi.org/10.1016/j.polymertesting.2012.05.011
» http://dx.doi.org/10.1016/j.polymertesting.2012.05.011 -
15Rana, A. K., Mandal, A., & Bandyopadhyay, S. (2003). Bandyopadhyay S.: Short jute fiber reinforced polypropylene composites: effect of compatibilizer, impact modifier and fiber loading. Composites Science and Technology, 63(6), 801-806. http://dx.doi.org/10.1016/S0266-3538(02)00267-1
» http://dx.doi.org/10.1016/S0266-3538(02)00267-1 -
16Rabello, M., & De Paoli, M.-A. (2013). Aditivação de termoplásticos. São Paulo: Artliber.
-
17Faker, M., Razavi Aghjeh, M. K., Ghaffari, M., & Seyyedi, S. A. (2008). Rheology, morphology and mechanical properties of polyethylene/ethylene vinyl acetate copolymer (PE/EVA) blends. European Polymer Journal, 44(6), 1834-1842. http://dx.doi.org/10.1016/j.eurpolymj.2008.04.002
» http://dx.doi.org/10.1016/j.eurpolymj.2008.04.002 -
18Khonakdar, H. A., Wagenknecht, U., Jafari, S. H., Hässler, R., & Eslami, H. (2004). Dynamic mechanical properties and morphology of polyethylene/ethylene vinyl acetate copolymer blends. Advances in Polymer Technology, 23(4), 307-315. http://dx.doi.org/10.1002/adv.20019
» http://dx.doi.org/10.1002/adv.20019 -
19Khonakdar, H. A., Jafari, S. H., Yavari, A., Asadinezhad, A., & Wagenknecht, U. (2005). Rheology, Morphology and estimation of interfacial tension of LDPE/EVA and HDPE/EVA blends. Polymer Bulletin, 54(1-2), 75-84. http://dx.doi.org/10.1007/s00289-005-0365-6
» http://dx.doi.org/10.1007/s00289-005-0365-6 -
20Araujo, J. R., Waldman, W. R., & De Paoli, M. A. (2008). Thermal properties of high density polyethylene composites with natural fibres: Coupling agent effect. Polymer Degradation & Stability, 93(10), 1770-1775. http://dx.doi.org/10.1016/j.polymdegradstab.2008.07.021
» http://dx.doi.org/10.1016/j.polymdegradstab.2008.07.021 -
21McNeill, I. C. (1997). Thermal degradation mechanisms of some addition polymers and copolymers. Journal of Analytical and Applied Pyrolysis, 40-41, 21-41. http://dx.doi.org/10.1016/S0165-2370(97)00006-5
» http://dx.doi.org/10.1016/S0165-2370(97)00006-5 -
22Ausias, G., Bourmaud, A., Coroller, G., & Baley, C. (2013). Study of the fibre morphology stability in polypropylene-flax composites. Polymer Degradation & Stability, 98(6), 1216-1224. http://dx.doi.org/10.1016/j.polymdegradstab.2013.03.006
» http://dx.doi.org/10.1016/j.polymdegradstab.2013.03.006 -
23Albano, C., Gonzalez, J., Ichazo, M., & Kaiser, D. (1999). Thermal stability of blends of polyolefins and sisal fiber. Polymer Degradation & Stability, 66(2), 179-190. http://dx.doi.org/10.1016/S0141-3910(99)00064-6
» http://dx.doi.org/10.1016/S0141-3910(99)00064-6
Publication Dates
-
Publication in this collection
07 June 2016 -
Date of issue
Apr-Jun 2016
History
-
Received
20 Feb 2015 -
Reviewed
14 Sept 2015 -
Accepted
27 Jan 2016










