Resumos
A leucemia promielocítica aguda (LPA) é um modelo da aplicabilidade clínica dos conhecimentos moleculares fisiopatológicos. Caracteriza-se por alterações genéticas recorrentes que envolvem o gene do receptor alfa do ácido retinóico. A conseqüência é uma proteína com sensibilidade reduzida ao ligante, com bloqueio da diferenciação mielóide. Entretanto, doses suprafisiológicas do ácido all-trans-retinóico (ATRA) são capazes de suplantar esta deficiência, e este é o princípio fundamental do tratamento da LPA, permitindo uma sobrevida livre de doença acima de 80% quando adequadamente tratada. Epidemiologicamente, difere dos demais subtipos de leucemia mielóide aguda por apresentar incidência predominante em adultos jovens e, aparentemente, maior incidência em países de colonização "latina". Contrastando com os excelentes resultados observados em países desenvolvidos, a mortalidade por LPA no Brasil ainda é alta, apesar da ampla disponibilidade das medicações no país.
Acute promyelocytic leukemia (APL) is a model of clinical applicability of the knowledge of molecular physiopathology. It is characterized by recurrent genetic involvement of the retinoic acid alpha receptor. The consequence is a protein with low sensibility to its ligand and a myeloid maturation arrest. However, higher doses of all-trans-retinoic acid (ATRA) are able to supersede this deficiency and this is the mainstay of APL treatment leading to over 80% disease free survival, when adequately treated. Epidemiologically, it differs from other acute myeloid leukemia due to a higher incidence in young adults and in countries of "Latin" colonization. Differing from excellent results observed in developed countries, APL mortality in Brazil is still high, despite the wide availability of drugs.
Acute myeloid leukemia; Acute promyelocytic leukemia
ARTIGO DE REVISÃO
Do paradigma molecular ao impacto no prognóstico: uma visão da leucemia promielocítica aguda
From the molecular model to the impact on prognosis: an overview on acute promyelocytic leukemia
Rafael Henriques Jácomo; Lorena Lobo de Figueiredo-Pontes; Eduardo Magalhães Rego* * Correspondência Laboratório de Hematologia Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto USP Av. Bandeirantes 3900, Monte Alegre, Ribeirão Preto SP CEP 14048-900 emrego@hcrp.fmrp.usp.br
RESUMO
A leucemia promielocítica aguda (LPA) é um modelo da aplicabilidade clínica dos conhecimentos moleculares fisiopatológicos. Caracteriza-se por alterações genéticas recorrentes que envolvem o gene do receptor alfa do ácido retinóico. A conseqüência é uma proteína com sensibilidade reduzida ao ligante, com bloqueio da diferenciação mielóide. Entretanto, doses suprafisiológicas do ácido all-trans-retinóico (ATRA) são capazes de suplantar esta deficiência, e este é o princípio fundamental do tratamento da LPA, permitindo uma sobrevida livre de doença acima de 80% quando adequadamente tratada. Epidemiologicamente, difere dos demais subtipos de leucemia mielóide aguda por apresentar incidência predominante em adultos jovens e, aparentemente, maior incidência em países de colonização "latina". Contrastando com os excelentes resultados observados em países desenvolvidos, a mortalidade por LPA no Brasil ainda é alta, apesar da ampla disponibilidade das medicações no país.
SUMMARY
Acute promyelocytic leukemia (APL) is a model of clinical applicability of the knowledge of molecular physiopathology. It is characterized by recurrent genetic involvement of the retinoic acid alpha receptor. The consequence is a protein with low sensibility to its ligand and a myeloid maturation arrest. However, higher doses of all-trans-retinoic acid (ATRA) are able to supersede this deficiency and this is the mainstay of APL treatment leading to over 80% disease free survival, when adequately treated. Epidemiologically, it differs from other acute myeloid leukemia due to a higher incidence in young adults and in countries of "Latin" colonization. Differing from excellent results observed in developed countries, APL mortality in Brazil is still high, despite the wide availability of drugs.
Key words: Acute myeloid leukemia. Acute promyelocytic leukemia.
INTRODUÇÃO
As leucemias mielóides agudas (LMA) constituem um grupo heterogêneo de doenças caracterizado pela expansão clonal de progenitores hematopoéticos imaturos na medula óssea levando ao bloqueio da hematopoese normal. Alterações genéticas foram identificadas na maioria dos casos de LMA, e a análise da genômica funcional levou à elaboração de modelos fisiopatológicos distintos para os subtipos de LMA, os quais por sua vez foram base para o desenvolvimento de novas abordagens terapêuticas. O paradigma desta evolução da pesquisa básico-clínica foi o subtipo associado à translocação entre os cromossomos 15 e 17 t(15;17) conhecido como leucemia promielocítica aguda (LPA).
A leucemia promielocítica aguda, ou LMA com translocação t(15;17), corresponde a cerca de 20% a 25% das LMAs nos países latino-americanos1-3 e apresenta características clínicas, morfológicas e biológicas peculiares, que a distinguem dos demais subtipos dessa doença. Do ponto de vista morfológico, corresponde ao subtipo M3 da classificação franco-américo-britânica (FAB) e caracteriza-se pela interrupção da maturação mielóide e acúmulo de células leucêmicas semelhantes a promielócitos na medula óssea4. Clinicamente, difere das outras LMAs por estar associada à coagulopatia em cerca de 60% a 90% dos pacientes5-7, e esta é a principal responsável pelas altas taxas de mortalidade precoce durante as fases iniciais do tratamento.
A LPA é o primeiro modelo de doença genética adquirida tratada com uma droga específica, o ácido all-trans-retinóico (ATRA), que induz diferenciação terminal seguida de apoptose das células leucêmicas. Atualmente, o tratamento adequado, incluindo a combinação de ATRA e antracíclicos, induz à remissão em mais de 90% dos pacientes e a sobrevida livre de doença em seis anos é superior a 80%8. Entretanto, o prognóstico depende do diagnóstico imediato e acurado, assim como da instituição precoce da terapêutica específica.
Bases moleculares
A t(15;17) detectada em mais de 90% dos casos de LPA é o resultado da quebra e fusão dos genes PML, localizado no cromossomo 15, e RARa, localizado no cromossomo 179 (Figura 1). A conseqüência é a produção de dois genes de fusão que codificam as proteínas híbridas PML-RARa e RARa-PML10-11. Os híbridos PML-RARa retêm a maioria dos domínios funcionais das proteínas parentais e atuam como produtos oncogênicos dominante-negativos, interferindo nas funções da via retinóide e do PML11-12.
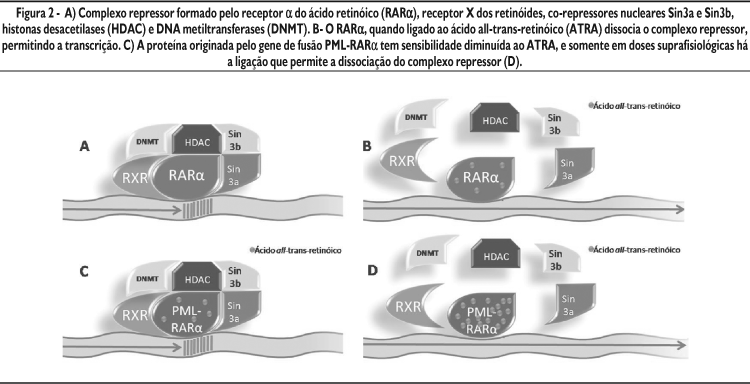
Os retinóides são derivados da vitamina A necessários a múltiplos processos biológicos13 e exercem papel fundamental na diferenciação mielóide. Sua ação depende de receptores intracelulares, os receptores do ácido retinóide, que existem como isoformas distintas distribuídas segundo padrões específicos em cada tecido. A isoforma a é a mais relevante para a mielopoese e, na presença de concentrações fisiológicas do ATRA, é capaz de associar-se ao seu ligante, migrar do citoplasma ao núcleo e dimerizar-se com uma segunda classe de receptores retinóides, os receptores X do ácido retinóico (RXR). Este complexo reconhece seqüências específicas de oligonucleotídeos presentes nas regiões promotoras de vários genes. Ao ligar-se aos co-repressores nucleares Sin3a e Sin3b (NCor) e, posteriormente, às histonas desacetilases (HDAC), leva à desacetilação das histonas, com conseqüente compactação da cromatina e repressão da transcrição gênica. O complexo repressor compreende, ainda, a DNA metiltransferase (DNMT), responsável pela metilação do DNA e inibição da transcrição (Figura 2). O gene de fusão PML-RARa gera uma oncoproteína com sensibilidade diminuída à ação dos retinóides14. Desta forma, concentrações fisiológicas do ácido retinóide não são capazes de dissociar o complexo repressor e o bloqueio da transcrição é mantido. Entre os genes regulados pelo complexo RARa/RXR/NCor/DNMT/HDAC estão genes importantes para a diferenciação mielóide, como os genes da elastase e mieloperoxidase. A oncoproteína PML-RARa leva, portanto, à interrupção da maturação mielóide no estágio de promielócito. Doses farmacológicas do ATRA são capazes de se ligar à PML-RARa e promover a dissociação do complexo, permitindo a progressão da transcrição15.
A proteína codificada pelo PML encontra-se organizada em dímeros e atua como supressor de tumor através do controle da apoptose e diferenciação celular16-17. A oncoproteína PML-RARa mantém a capacidade de ligação à proteína parental, porém desloca o PML de seu sítio subcelular normal, estruturas nucleares conhecidas como nuclear bodies (NBs), e interfere de forma dominante negativa na função da proteína nativa.
Em raros casos de LPA, o RARa está fusionado a outros parceiros: o PLZF (do inglês, Promyelocytic Leukemia Zinc Finger), NPM (do inglês, Nucleophosmin), NuMA (do inglês, Nuclear Mitotic Apparatus) e Stat5b (do inglês, Signal Transducer and Activator of Transcription), decorrentes de translocações entre o cromossomo 17 e os cromossomos 11, 5, 11 e 17, respectivamente18. Essas translocações variantes levam à formação de proteínas de fusão conhecidas genericamente como X/RARa, com sensibilidade diferenciada aos retinóides. Assim, pacientes com LPA resistente ao tratamento como ATRA podem ser portadores da t(11;17) PLZF/RARa. Esta proteína de fusão forma complexos repressores da transcrição não só através do domínio RARa, mas também do PLZF, o qual não exibe afinidade ao ATRA.
Apesar da clara correlação entre a t(15;17) e a doença, esta não é suficiente para, isoladamente, desencadear a LPA. Apenas 10% dos camundongos transgênicos que expressam o gene de fusão PML-RARa desenvolvem uma doença semelhante à LPA humana19. Outras alterações moleculares, tais como a presença do rearranjo FLT3-ITD ou uma desregulação da via do CEBPa, contribuem para o processo de leucemogênese e, conseqüentemente, para a ocorrência da manifestação clínica20-21.
Epidemiologia
Diferentemente dos outros subtipos de LMA, a LPA ocorre com maior freqüência em adultos jovens e tem incidência praticamente estável entre os 20 e 59 anos de idade. Não há predomínio de nenhum dos sexos e não está associada à síndrome mielodisplásica prévia. Existem raros relatos da doença secundária a quimioterapia, porém, ao contrário dos outros subtipos de LMA, não há piora do prognóstico com o tratamento adequado22.
A freqüência de LPA entre as LMAs é muito variável. Na maioria dos países desenvolvidos a taxa de incidência varia de 4% a 15%22, enquanto, em países de colonização latina e algumas regiões da Espanha, esta é de 20% a 28%1-3,23,24. Porém, a real incidência da LPA não é conhecida, e a maioria dos estudos baseia-se em registros hospitalares. Existe, ainda, uma variabilidade genética correlacionada à distribuição geográfica, com diferentes freqüências das isoformas do PML-RARa conforme a população estudada25-27.
Manifestações clínicas
Assim como nas outras LMAs, sintomas constitucionais como febre, astenia, hiporexia e perda ponderal predominam. Organomegalia, adenomegalia e infiltração leucêmica da pele ou sistema nervoso central são incomuns ao diagnóstico. Manifestações secundárias a leucostase são raras, pois a doença usualmente apresenta- se com leucopenia ou discreta leucocitose. Caracteristicamente, há uma propensão a sangramento aumentada, desproporcional à plaquetopenia. Os eventos hemorrágicos estão presentes em praticamente 60% dos pacientes ao diagnóstico, e a sua incidência tende a aumentar nos primeiros dias do tratamento. A coagulopatia é o principal fator adverso na evolução da doença, sendo responsável pelo maior número de óbitos no período inicial de tratamento5-7,28-33.
Esta coagulopatia depende de três fatores: ativação da cascata da coagulação, aumento da fibrinólise e proteólise. Os blastos da LPA expressam o fator tecidual34-35, ativador da via clássica da cascata da coagulação, e também induzem a produção de interleucina-136, aumentando a atividade pró-coagulante em células endoteliais. Ademais, um fator pró-coagulante alternativo (denominado Cancer Procoagulant) é liberado pelos blastos da LPA e ativa diretamente o fator X37. Isto leva à geração de fibrina de forma semelhante ao processo de coagulação intravascular disseminada (CIVD). Entretanto, ao contrário do que ocorre na CIVD, a vida média das plaquetas, bem como a concentração dos anticoagulantes naturais (antitrombina e proteína C) no plasma, encontram-se normais38-40.
O estado de hiperfibrinólise é evidenciado pelos baixos níveis de antifibrinolíticos, como o inibidor a2 da plasmina (a2PI)41 e o inibidor da ativação do plasminogênio (PAI-1) e por um déficit acentuado de fibrinogênio42. Além disso, embora a concentração do inibidor da fibrinólise ativado pela trombina (TAFI) seja normal, sua atividade apresenta-se acentuadamente reduzida43. Tais alterações resultam em um quadro compatível com o aumento da produção de plasmina. Expressão aumentada de anexina II foi detectada em células leucêmicas de LPA44. Esta proteína é um receptor de superfície celular para o plasminogênio bem como para um dos seus ativadores, o ativador tecidual do plasminogênio (t-PA). Portanto, a superexpressão da anexina II deve participar da fisiopatogenia do estado de hiperfibrinólise característico da coagulopatia da LPA.
Outros fatores estão associados à coagulopatia, tais como: a) aumento dos níveis séricos de elastase45 que, além de agir diretamente na destruição do coágulo de fibrina, também tem ação inibitória sobre a coagulação mediante a lise de fibrina e geração de seus produtos de degradação, que têm ação inibitória sobre a coagulação e; b) aumento das concentrações do fator de von Willebrand e dos seus fragmentos em circulação46, estes últimos provavelmente como conseqüência da ação proteolítica da plasmina e também da elastase.
Diagnóstico laboratorial
O hemograma geralmente evidencia pancitopenia e quando há leucocitose, esta costuma ser discreta. Blastos com morfologia de promielócitos, assim como evidências de anemia microangiopática (esquizócitos), podem ser vistos na lâmina de sangue periférico. O mielograma evidencia infiltração maciça por promielócitos neoplásicos, que se coram fortemente à reação da mieloperoxidase e ao Sudan Black. Dois subtipos morfológicos podem ocorrer: o padrão hipergranular (ou subtipo FAB M3 clássico) e o microgranular (ou subtipo FAB M3 variante ou hipogranular), este último correspondendo a 25% dos casos. A forma clássica se caracteriza pela presença de promielócitos anormais com granulação azurofílica abundante, núcleo irregular e bastonetes de Auer, que freqüentemente se organizam em feixes, caracterizando as chamadas células de Faggot. Na variante hipogranular, geralmente associada à leucocitose, observa-se grande número de blastos com núcleos bilobulados ou reniformes e granulação citoplasmática discreta. Pode haver a coexistência de blastos hipergranulares típicos em menor número. Há ainda uma forma hiperbasofílica, rara, na qual as células leucêmicas têm alta relação núcleo-citoplasmática e citoplasma fortemente basofílico com granulação esparsa ou ausente47.
O estudo de imunofenotipagem evidencia blastos com elevada auto-fluorescência, expressando marcadores mielóides precoces como o CD117 com baixa intensidade de fluorescência, CD13 com padrão homogêneo de intensidade de fluorescência e CD33 com padrão heterogêneo. O marcador de células precursoras hematopoéticas CD34 é usualmente negativo, assim como HLA-DR. Ao contrário do que ocorre em promielócitos normais, os marcadores indicativos de maturidade mielóide CD11b e CD15 são negativos ou de muito baixa expressão. Por ser um método rápido, a imunofenotipagem reforça a suspeita diagnóstica de LPA e ajuda na indicação terapêutica precoce, mas não é um método adequado para o diagnóstico definitivo48.
A confirmação diagnóstica deve ser feita por técnicas capazes de detectar a t(15;17) ou o gene híbrido PML-RARa. Citogenética convencional, hibridização por sondas de fluorescência in situ (FISH, do inglês Fluorescence in situ hybridization) e reação em cadeia de polimerase por transcriptase reversa (RT-PCR) são opções disponíveis. A técnica de FISH tem menor sensibilidade que a RT-PCR, porém é mais específica. O cariótipo convencional tem a vantagem de permitir o diagnóstico de alterações citogenéticas adicionais, porém o tempo de execução é maior e nem sempre se observam metáfases em número e qualidade necessárias para análise. Outra opção atualmente disponível é a imunofluorescência com anticorpos anti-PML (PGM3). Este anticorpo se liga à proteína PML que se localiza em estruturas denominadas nuclear bodies. Nos pacientes portadores do rearranjo PML-RARa, a PML encontra-se redistribuída e observa-se, à microscopia, um padrão microparticulado49 (Figura 2). A imunofluorescência é útil para o rápido diagnóstico, particularmente onde não se dispõe de métodos moleculares, porém não deve substituir a confirmação molecular.
Fatores prognósticos ao diagnóstico
Vários fatores prognósticos foram identificados na LPA. A elevada contagem inicial de glóbulos brancos parece estar associada a maior mortalidade durante a indução e maior incidência de recaída. Embora o valor limite para a definição de pior prognóstico seja variável entre os estudos, a maior parte dos grupos adotam o valor de 10.000 leucócitos/µL8,50-51. A idade avançada também está associada ao prognóstico devido a maior mortalidade precoce bem como intolerância à quimioterapia. Em pacientes com idade maior ou igual a 70 anos, vários protocolos recomendam a redução da dose de antracíclicos. A importância prognóstica de anormalidades cromossômicas adicionais à t(15;17) não está completamente estabelecida, sendo que, na maioria dos estudos, a presença dessas alterações não influencia o prognóstico52.
Os grupos PETHEMA (espanhol) e GIMEMA (italiano) observaram duas variáveis que, independentemente, foram capazes de prever recaída. Os pacientes que apresentavam contagem plaquetária maior que 40 x 103/µL apresentavam menor taxa de recaída, tanto molecular quanto hematológica. O mesmo foi observado para pacientes com menos que 10 x 103/µL leucócitos. Com base nestes valores, três grupos de risco foram estabelecidos (Tabela 1) e uma estratégia de tratamento intensificada para os de pior prognóstico foi elaborada51.
Tratamento específico
O tratamento da LPA sofreu importantes modificações nos últimos 20 anos e difere dos esquemas utilizados para as demais LMAs. O maior impacto no tratamento da LPA foi, sem dúvida, a demonstração de que o ácido all-Trans-Retinóico (ATRA), em doses farmacológicas, permite a progressão da diferenciação celular53. Desta forma, o clone leucêmico progride na maturação mielóide, tornando-se susceptível aos mecanismos de morte celular. O tratamento com ATRA deve ser iniciado imediatamente diante da suspeita morfológica, mesmo antes da confirmação genética do diagnóstico, pois leva à melhora da coagulopatia e diminuição do risco de sangramento grave28. Apesar de seu uso como monoterapia levar à remissão hematológica, todos os pacientes apresentam recaída. Assim, exceto para os pacientes com alguma contra-indicação clínica ao uso de antracíclicos, a indução deve consistir da sua administração associada ao ATRA.
Com o esquema combinado (ATRA e antracíclico), alcança-se remissão molecular em até 99% dos pacientes, com sobrevida livre de doença de 90% aos cinco anos do diagnóstico6,8,51,54-57. O tratamento é usualmente dividido em três fases: indução de remissão, consolidação e manutenção. As duas primeiras são fundamentadas no uso de ATRA e algum antracíclico, enquanto que a última é composta de ciclos de ATRA associado a metotrexate e mercaptopurina em baixas doses. O grupo PETHEMA propôs um escalonamento da dose de antracíclico na consolidação conforme o grupo de risco do paciente, uma estratégia que visa a diminuir a taxa de recaída nos grupos de risco alto e intermediário8. Entretanto, os pacientes classificados como alto risco ainda mantêm taxas de recaída acima de 20%, e o novo protocolo deste grupo (LPA2005) associa a citarabina ao primeiro e terceiro ciclos da consolidação para estes pacientes.
Apesar da excelente resposta ao ATRA em pacientes com LPA associada ao PML-RARa, os portadores de outras translocações envolvendo o RARa apresentam sensibilidade variável a esta medicação. Nestes grupos, outras opções devem ser buscadas para o tratamento, como o arsênico e outros agentes diferenciadores.
Síndrome de diferenciação
O uso do ATRA está associado, em 5% a 20% dos pacientes, a um quadro conhecido como síndrome ATRA, síndrome retinóide ou síndrome de diferenciação, potencialmente fatal58. As manifestações clínicas são bastante variadas, mas predominam sintomas respiratórios, associados ou não a edema ou infiltrados pulmonares. Febre, dor óssea, ganho de peso, edema, falência cardíaca, insuficiência renal, derrames cavitários, cefaléia (que deve ser diferenciada da síndrome de pseudotumor cerebral) e síndrome de Sweet são outros achados observados, comumente precedidos por elevação da contagem de leucócitos. O quadro inicia-se em geral entre o terceiro e o décimo quarto dia de tratamento com ATRA, mas há relatos de ocorrência após a primeira dose59.
A etiologia desta síndrome não é completamente entendida, porém, a infiltração de diferentes órgãos por células mielóides é um achado invariável. Provavelmente, a expressão diferenciada de moléculas de adesão como a ICAM-1 e a produção de citocinas, como IL-8 e MMP-9 estão envolvidas. A via final é a lesão endotelial, inflamação e sangramento no tecido acometido. A expressão aumentada do CD13 e a hiperleucocitose estão, aparentemente, associadas a maior incidência da síndrome. Por outro lado, esta é menos freqüente no subtipo morfológico clássico e nos pacientes tratados com antracíclico concomitante ao ATRA na indução59.
A rápida identificação dos sintomas e o início precoce do tratamento é essencial, pois a síndrome retinóide leva à diminuição da sobrevida livre de eventos e da sobrevida global6,51,55,58,60. O tratamento consiste na interrupção temporária do tratamento com ATRA e associação de dexametasona na dose de 10 mg de 12 em 12 horas por pelo menos três a quatro dias até resolução dos sintomas. Além disso, o suporte clínico com diuréticos e ventilação mecânica pode ser necessário. Apesar da boa resposta ao uso de corticosteróides, o uso profilático de prednisona não mostrou redução da morbimortalidade associada à síndrome retinóide.
Medidas de controle da coagulopatia
O tratamento da LPA envolve não apenas a escolha correta do esquema quimioterápico, mas também um suporte hemoterápico agressivo. Devido ao risco de sangramento, a contagem de plaquetas deve ser mantida acima de 30 x 103/µL. Plasma fresco congelado e crioprecipitado devem ser prescritos enquanto houver evidência clínica ou laboratorial de consumo de fatores da coagulação (avaliado por quantificação de Dímeros D, Produtos de Degradação da Fibrina e fibrinogênio bem como determinação do Tempo de Protrombina e Tempo de Tromboplastina Parcial ativada), objetivando manter o fibrinogênio acima de 150 mg/µL. Pacientes que apresentam sangramento ativo ou risco hemorrágico elevado (idosos, creatinina elevada e hiperleucocitose) devem ser acompanhados com maior atenção.
As evidências da ativação da cascata da coagulação podem sugerir que o uso de anticoagulantes como a heparina ou antifibrinolíticos levaria ao bloqueio da coagulopatia. Entretanto, os estudos clínicos até o momento disponíveis não evidenciaram diminuição de sangramento e seu uso não é recomendado.
Monitorização do tratamento
Após a indução de remissão, havendo recuperação das contagens hematológicas no sangue periférico (contagem de neutrófilos acima de 1,5 x 103/µL e plaquetas acima de 50 x 103/µL) deve-se realizar punção de medula óssea para mielograma. Nesta fase, pode haver um padrão de hipercelularidade e retardo de maturação na medula óssea, que não deve ser confundido com refratariedade. Nestes casos, recomenda-se a repetição do mielograma semanalmente e continuar o tratamento com ATRA até a obtenção de resposta hematológica completa ou por até 90 dias. Ademais, os resultados de RT-PCR, cariótipo ou FISH após a indução não têm valor prognóstico e, portanto, não devem modificar a terapia.
Remissão molecular é atingida em 90% a 99% dos pacientes que recebem dois a três ciclos de consolidação, incluindo um antracíclico6,8,51,55-57. Após a terceira consolidação, nova amostra de medula óssea deve ser colhida, para mielograma e RT-PCR. Ao contrário do esperado após a indução, nesta fase do tratamento, o resultado do RT-PCR com técnicas de baixa sensibilidade (10-3 e 10-4) para o PML-RARa é capaz de predizer o risco de recaída precoce. Os pacientes que persistem com o transcrito PML-RARa têm pior prognóstico61 e devem ser submetidos a novas estratégias como o uso do anticorpo monoclonal anti-CD33 ligado a quimioterápico (gentuzumab ozogamicina) e/ou do trióxido de arsênico seguidas de transplante de medula óssea (TMO). Os demais devem prosseguir à manutenção.
Alternativas terapêuticas na recaída
A recaída hematológica segue os mesmos critérios do diagnóstico. A recaída ou persistência molecular, por sua vez, é considerada na presença de dois resultados positivos consecutivos do RT-PCR para o PML-RARa obtidos com intervalo mínimo de duas semanas.
Cerca de 10% dos pacientes tratados com ATRA associado a antracíclico apresentam recaída hematológica6,8,57,61, sendo raras as recaídas extramedulares. Os pacientes em recaída que receberam ATRA há menos de seis meses devem ser tratados com esquemas alternativos. Apesar de praticamente 100% dos pacientes que não receberam ATRA previamente responderem a esta droga após a recaída, apenas 20% dos pacientes previamente expostos entram em remissão.
Atualmente, a alternativa mais utilizada para os casos de recaída é o tratamento com trióxido de arsênico (ATO). Esta medicação é conhecida há mais de 2.400 anos pelos chineses e gregos, porém apenas recentemente a sua utilidade no tratamento de neoplasias foi descoberta. Em altas doses, leva à apoptose, enquanto baixas doses promovem a diferenciação celular. A droga tem três mecanismos de ação principais: 1) produção de produtos reativos do oxigênio que induzem a fosforilação e ativação da via da Jun N-terminal kinase (JNK), desencadeando apoptose; 2) a fosforilação e sumolização do PML-RARa levando à sua degradação; e 3) inibição da transcrição do hTERT e conseqüente diminuição da atividade da telomerase, levando à fusão cromossômica e apoptose62.
O ATO é administrado por via intravenosa e apresenta meia vida de 30 dias, com baixa toxicidade se comparado à quimioterapia convencional, representada por desconforto gastrointestinal, elevação de enzimas hepáticas, neuropatia, hiperglicemia, arritmias cardíacas e síndrome de diferenciação semelhante à síndrome ATRA. O esquema para tratamento da recaída inclui um primeiro ciclo de indução com 0.15 mg/kg/dia até obtenção de remissão completa ou 50 dias de tratamento, seguido de dois ciclos de consolidação com a mesma dose de ATO por cinco dias durante cinco semanas. A taxa de remissão molecular obtida com a terapia isolada é de aproximadamente 86%, com sobrevida de 77% em três anos63. A complementação do tratamento pode ser feita com TMO alogênico para os pacientes com doador HLA idêntico ou autólogo, sendo neste último caso mandatória a negatividade do RT-PCR antes do procedimento.
Estudos preliminares com o anticorpo monoclonal gentuzumab ozogamicina também mostram boas respostas nos casos de recaída64.
A LPA no Brasil
Dados sobre a epidemiologia da LPA no Brasil são escassos e baseiam-se em registros hospitalares. Ademais, as bases de dados não registram separadamente os subtipos de leucemias mielóides agudas. Um recente levantamento realizado em 12 centros que tratam neoplasias hematológicas contabilizou 157 pacientes, correspondendo a 28% dos casos de LMAs65. A percentagem de pacientes classificados como alto risco foi de 36,9%, significativamente maior do que aquela relatada em estudos clínicos de países desenvolvidos. Embora esta apresentação possa ser uma característica desta população, não se pode descartar a hipótese de ser devida à dificuldade de acesso dos pacientes a um serviço especializado. O fato que mais chamou atenção, entretanto, foi a sobrevida global em três anos, de 56,3%, inferior à relatada na literatura. A principal causa de mortalidade foram as complicações hemorrágicas nos primeiros 14 dias de tratamento. De fato, 26,4% dos pacientes morreram neste período, sendo 89% em decorrência de manifestações hemorrágicas
As diferenças entre os dados apresentados pelos grupos estrangeiros e os dados locais tornam-se ainda mais contundentes se lembrarmos que tanto os antracíclicos (especificamente a daunorrubicina e o mitoxantrone) quanto o ATRA estão disponíveis na rede de atendimento do Sistema Único de Saúde. Ademais, boa parte dos centros utiliza esquemas terapêuticos semelhantes aos sugeridos pelos grupos PETHEMA e GIMEMA. Fica claro, portanto, que não basta a disponibilidade das medicações para que consigamos resultados pelo menos comparáveis aos dos países desenvolvidos. Primeiramente, é necessário que se crie uma rede de cooperação entre os centros, semelhante à existente em outros países. De forma complementar, é necessário disponibilizar aos hematologistas a possibilidade de trocar experiências com colegas do país e com especialistas do exterior.
O consórcio internacional em leucemia promielocítica aguda
O IC-APL (International Consortium on Acute Promyeolocytic Leukemia) foi criado em 2005 por integrantes do Comitê de Membros Internacionais da Sociedade de Hematologia Americana (IMC-ASH) e hematologistas brasileiros, mexicanos e jordanianos. Este grupo tem por objetivo implantar uma rede de cooperação entre centros nacionais e internacionais para melhorar o tratamento oferecido aos pacientes. Foi elaborado um protocolo de tratamento, baseado no protocolo LPA2005 do PETHEMA, adaptado à realidade destes países. A grande mudança foi a substituição da idarrubicina (um antracíclico de preço elevado e indisponível no México e em alguns centros brasileiros) pela daunorrubicina, de valor mais acessível e amplamente utilizado no tratamento de outras leucemias.
Um laboratório central é responsável pelos exames de RT-PCR e monitoramento da resposta molecular. Os pacientes são registrados por meio de uma ferramenta on-line, que permite a inserção das diversas informações do tratamento e o compartilhamento destas com outros centros. Possibilita, também, a utilização fácil e ampla, uma vez que o único requisito é um computador com acesso à internet. O grupo reúne-se semanalmente por teleconferência em um software desenvolvido pelo St. Jude Children's Hospital, quando são discutidas questões referentes aos dados clínicos e evolução dos pacientes. Uma vez por mês há reuniões on-line com especialistas da Espanha, Itália, Alemanha, Holanda e Estados Unidos para discussão clínica e troca de informações a respeito dos exames laboratoriais.
Perspectivas e Conclusões
O tratamento da LPA tem como característica peculiar, além do direcionamento ao alvo molecular, a baixa toxicidade, quando comparado às outras LMAs. Acredita-se ainda ser possível reduzi-la com novas estratégias terapêuticas. O uso do trióxido de arsênico em pacientes recém-diagnosticados, por exemplo, é uma alternativa com alta taxa de remissão. Há relatos de uso do ATRA em combinação com arsênico e taxas de remissão semelhantes ou mesmo superiores às observadas nos estudos consagrados66. Uma conseqüência temida desta associação, a hiperleucocitose, tem sido combatida com a associação de algum quimioterápico quando a contagem de leucócitos ultrapassa 10 x 103/µL. Isso significa que alguns pacientes poderiam ser tratados sem a utilização de quimioterápicos convencionais. Estão em avaliação estratégias como o uso de gentuzumab ozogamicina como primeira linha, doses reduzidas de idarrubicina e o HuM195. Entretanto, qualquer mudança no tratamento de uma doença que apresenta taxas de cura tão altas com o esquema atual deve ser vista com cautela. Estas novas terapêuticas devem ficar reservadas para estudos clínicos investigacionais bem como para pacientes que não podem ser submetidos ou se recusam à quimioterapia.
A LPA tem o melhor prognóstico entre as leucemias agudas dos adultos. Os esquemas quimioterápicos bem como as orientações de seguimento já são bem estabelecidos na literatura, e acessíveis para o médico habituado ao tratamento antineoplásico. O correto diagnóstico, associado à pronta instituição do tratamento, assim como a detecção precoce das complicações são essenciais para que possamos alcançar níveis de resposta semelhantes aos obtidos por grupos estrangeiros. A iniciativa do IC-APL provavelmente contribuirá para a melhora dos resultados, uma vez que existem, no país, mão-de-obra médica treinada e disponibilidade das medicações.
Conflito de interesse: não há
Artigo recebido: 25/07/07
Aceito para publicação: 06/11/07
Trabalho realizado no Laboratório de Hematologia Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto, SP
- 1. Douer D, Preston-Martin S, Chang E, Nichols PW, Watkins KJ, Levine AM. High frequency of acute promyelocytic leukemia among Latinos with acute myeloid leukemia. Blood. 1996;87(1):308-13.
- 2. Mendes WL, Coser VM, Ramos G, Pereira W, Lopes LF, de Oliveira MS. The apparent excess of acute promyelocytic leukemia in infant acute leukemias in Brazil. Haematologica. 2004;89(11):ELT16.
- 3. Otero JC, Santillana S, Fereyros G. High frequency of acute promyelocytic leukemia among Latinos with acute myeloid leukemia. Blood. 1996;88(1):377.
- 4. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33(4):451-8.
- 5. Candoni A, Damiani D, Michelutti A, Masolini P, Michieli M, Michelutti T, et al. Clinical characteristics, prognostic factors and multidrug-resistance related protein expression in 36 adult patients with acute promyelocytic leukemia. Eur J Haematol. 2003;71(1):1-8.
- 6. Sanz MA, Martin G, Rayon C, Esteve J, Gonzalez M, az-Mediavilla J, et al. A modified AIDA protocol with anthracycline-based consolidation results in high antileukemic efficacy and reduced toxicity in newly diagnosed PML/RARalpha-positive acute promyelocytic leukemia. PETHEMA group. Blood. 1999;94(9):3015-21.
- 7. Hernandez JM, Martin G, Gutierrez NC, Cervera J, Ferro MT, Calasanz MJ, et al. Additional cytogenetic changes do not influence the outcome of patients with newly diagnosed acute promyelocytic leukemia treated with an ATRA plus anthracyclin based protocol. A report of the Spanish group PETHEMA. Haematologica. 2001;86(8):807-13.
- 8. Sanz MA, Martin G, Gonzalez M, Leon A, Rayon C, Rivas C, et al. Risk-adapted treatment of acute promyelocytic leukemia with all-trans-retinoic acid and anthracycline monochemotherapy: a multicenter study by the PETHEMA group. Blood. 2004;103(4):1237-43.
- 9. Rowley JD, Golomb HM, Vardiman J, Fukuhara S, Dougherty C, Potter D. Further evidence for a non-random chromosomal abnormality in acute promyelocytic leukemia. Int J Cancer. 1977;20(6):869-72.
- 10. Goddard AD, Borrow J, Solomon E. A previously uncharacterized gene, PML, is fused to the retinoic acid receptor alpha gene in acute promyelocytic leukaemia. Leukemia. 1992;6(Suppl 3):117S-9S.
- 11. Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93(10):3167-215.
- 12. Perez A, Kastner P, Sethi S, Lutz Y, Reibel C, Chambon P. PMLRAR homodimers: distinct DNA binding properties and heteromeric interactions with RXR. EMBO J. 1993;12(8):3171-82.
- 13. Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, et al. International Union of Pharmacology. LX. Retinoic acid receptors. Pharmacol Rev. 2006;58(4):712-25.
- 14. Grignani F, Ferrucci PF, Testa U, Talamo G, Fagioli M, Alcalay M, et al. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell. 1993;74(3):423-31.
- 15. Muindi J, Frankel SR, Miller WH, Jr., Jakubowski A, Scheinberg DA, Young CW, et al. Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: implications for relapse and retinoid "resistance" in patients with acute promyelocytic leukemia. Blood. 1992;79(2):299-303.
- 16. Rego EM, Wang ZG, Peruzzi D, He LZ, Cordon-Cardo C, Pandolfi PP. Role of promyelocytic leukemia (PML) protein in tumor suppression. J Exp Med. 200119;193(4):521-9.
- 17. Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, et al. PML is essential for multiple apoptotic pathways. Nat Genet. 1998;20(3):266-72.
- 18. Redner RL. Variations on a theme: the alternate translocations in APL. Leukemia. 2002;16(10):1927-32.
- 19. Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood. 1997;89(2):376-87.
- 20. Truong BT, Lee YJ, Lodie TA, Park DJ, Perrotti D, Watanabe N, et al. CCAAT/Enhancer binding proteins repress the leukemic phenotype of acute myeloid leukemia. Blood. 2003;101(3):1141-8.
- 21. Yoo SJ, Park CJ, Jang S, Seo EJ, Lee KH, Chi HS. Inferior prognostic outcome in acute promyelocytic leukemia with alterations of FLT3 gene. Leuk Lymphoma. 2006;47(9):1788-93.
- 22. Douer D. The epidemiology of acute promyelocytic leukaemia. Best Pract Res Clin Haematol. 2003;16(3):357-67.
- 23. Sierra M, Alonso A, Odero MD, Gonzalez MB, Lahortiga I, Perez JJ, et al. Geographic differences in the incidence of cytogenetic abnormalities of acute myelogenous leukemia (AML) in Spain. Leuk Res. 2006;30(8):943-8.
- 24. Tomas JF, Fernandez-Ranada JM. About the increased frequency of acute promyelocytic leukemia among Latinos: the experience from a center in Spain. Blood. 1996;88(6):2357-8.
- 25. Chauffaille ML, Figueiredo MS, Beltrani R, Antunes SV, Yamamoto M, Kerbauy J. Acute promyelocytic leukemia: the study of t(15;17) translocation by fluorescent in situ hybridization, reverse transcriptase-polymerase chain reaction and cytogenetic techniques. Braz J Med Biol Res. 2001;34(6):735-43.
- 26. Melo RA, de Vasconcellos JF, Melo FC, Machado CG, Lacerda TM, Souto FR. PML-RARalpha fusion gene transcripts and biological features in acute promyelocytic leukemia patients. Clin Lab Haematol. 2006;28(2):126-9.
- 27. Ruiz-Arguelles GJ, Garces-Eisele J, Reyes-Nunez V, Gomez-Rangel JD, Ruiz-Delgado GJ. More on geographic hematology: the breakpoint cluster regions of the PML/RARalpha fusion gene in Mexican Mestizo patients with promyelocytic leukemia are different from those in Caucasians. Leuk Lymphoma. 2004;45(7):1365-8.
- 28. Di BE, Avvisati G, Castaman G, Luce VM, De S, V, Rodeghiero F, et al. Early haemorrhagic morbidity and mortality during remission induction with or without all-trans retinoic acid in acute promyelocytic leukaemia. Br J Haematol. 2000;108(4):689-95.
- 29. Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337(15):1021-8.
- 30. Steven L Soignet, Peter G Maslak. Acute promyelocytic leukemia. In: Greer JP, Forester J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe's clinical hematology. 11th ed. Philadelphia: Lippincott Williams & Wilkins; 2004. p. 2191-205.
- 31. Cordonnier C, Vernant JP, Brun B, Heilmann MG, Kuentz M, Bierling P, et al. Acute promyelocytic leukemia in 57 previously untreated patients. Cancer. 1985;55(1):18-25.
- 32. Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337(15):1021-8.
- 33. Cunningham I, Gee TS, Reich LM, Kempin SJ, Naval AN, Clarkson BD. Acute promyelocytic leukemia: treatment results during a decade at Memorial Hospital. Blood. 1989;73(5):1116-22.
- 34. Koyama T, Hirosawa S, Kawamata N, Tohda S, Aoki N. All-trans retinoic acid upregulates thrombomodulin and downregulates tissue-factor expression in acute promyelocytic leukemia cells: distinct expression of thrombomodulin and tissue factor in human leukemic cells. Blood. 1994;84(9):3001-9.
- 35. Zhu J, Guo WM, Yao YY, Zhao WL, Pan L, Cai X, et al. Tissue factors on acute promyelocytic leukemia and endothelial cells are differently regulated by retinoic acid, arsenic trioxide and chemotherapeutic agents. Leukemia. 1999;13(7):1062-70.
- 36. Cozzolino F, Torcia M, Miliani A, Carossino AM, Giordani R, Cinotti S, et al. Potential role of interleukin-1 as the trigger for diffuse intravascular coagulation in acute nonlymphoblastic leukemia. Am J Med. 1988;84(2):240-50.
- 37. Falanga A, Alessio MG, Donati MB, Barbui T. A new procoagulant in acute leukemia. Blood. 1988;71(4):870-5.
- 38. Bennett M, Parker AC, Ludlam CA. Platelet and fibrinogen survival in acute promyelocytic leukaemia. Br Med J1976;2(6035):565.
- 39. Zhu J, Guo WM, Yao YY, Zhao WL, Pan L, Cai X, et al. Tissue factors on acute promyelocytic leukemia and endothelial cells are differently regulated by retinoic acid, arsenic trioxide and chemotherapeutic agents. Leukemia. 1999;13(7):1062-70.
- 40. Sakata Y, Murakami T, Noro A, Mori K, Matsuda M. The specific activity of plasminogen activator inhibitor-1 in disseminated intravascular coagulation with acute promyelocytic leukemia. Blood. 1991;77(9):1949-57.
- 41. Sakata Y, Murakami T, Noro A, Mori K, Matsuda M. The specific activity of plasminogen activator inhibitor-1 in disseminated intravascular coagulation with acute promyelocytic leukemia. Blood. 1991;77(9):1949-57.
- 42. Aoki N, Moroi M, Matsuda M, Tachiya K. The behavior of alpha2-plasmin inhibitor in fibrinolytic states. J Clin Invest. 1977;60(2):361-9.
- 43. Meijers JC, Oudijk EJ, Mosnier LO, Bos R, Bouma BN, Nieuwenhuis HK, et al. Reduced activity of TAFI (thrombin-activatable fibrinolysis inhibitor) in acute promyelocytic leukaemia. Br J Haematol. 2000;108(3):518-23.
- 44. Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, Hajjar KA. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med. 1999;340(13):994-1004.
- 45. Sterrenberg L, Haak HL, Brommer EJ, Nieuwenhuizen W. Evidence of fibrinogen breakdown by leukocyte enzymes in a patient with acute promyelocytic leukemia. Haemostasis. 1985;15(2):126-33.
- 46. Federici AB, Falanga A, Lattuada A, Di RN, Barbui T, Mannucci PM. Proteolysis of von Willebrand factor is decreased in acute promyelocytic leukaemia by treatment with all-trans-retinoic acid. Br J Haematol. 1996;92(3):733-9.
- 47. Brunning RD, Matutes E, Flandrin G, Vardiman J, Bennet J, Head D, et al. Acute myeloid leukemia with recurrent genetic abnormalities. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. Pathology & genetics tumors of haematopoietic and lymphoid tissues. Geneve: WHO; 2001. p.81-91.
- 48. Orfao A, Chillon MC, Bortoluci AM, Lopez-Berges MC, Garcia-Sanz R, Gonzalez M, et al. The flow cytometric pattern of CD34, CD15 and CD13 expression in acute myeloblastic leukemia is highly characteristic of the presence of PML-RARalpha gene rearrangements. Haematologica. 1999;84(5):405-12.
- 49. Gomis F, Sanz J, Sempere A, Plume G, Senent ML, Perez ML, et al. Immunofluorescent analysis with the anti-PML monoclonal antibody PG-M3 for rapid and accurate genetic diagnosis of acute promyelocytic leukemia. Ann Hematol. 2004;83(11):687-90.
- 50. Asou N, Adachi K, Tamura U, Kanamaru A, Kageyama S, Hiraoka A, et al. Analysis of prognostic factors in newly diagnosed patients with acute promyelocytic leukemia: the APL92 study of the Japan Adult Leukemia Study Group (JALSG). Cancer Chemother Pharmacol. 2001;48(Suppl 1):S65-S71.
- 51. Sanz MA, Lo CF, Martin G, Avvisati G, Rayon C, Barbui T, et al. Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups. Blood. 2000;96(4):1247-53.
- 52. de BS, Chevret S, Sanz M, Dombret H, Thomas X, Guerci A, et al. Additional chromosomal abnormalities in patients with acute promyelocytic leukaemia (APL) do not confer poor prognosis: results of APL 93 trial. Br J Haematol. 2000;111(3):801-6.
- 53. Castaigne S, Chomienne C, Daniel MT, Ballerini P, Berger R, Fenaux P, et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76(9):1704-9.
- 54. Visani G, Gugliotta L, Tosi P, Catani L, Vianelli N, Martinelli G, et al. All-trans retinoic acid significantly reduces the incidence of early hemorrhagic death during induction therapy of acute promyelocytic leukemia. Eur J Haematol. 2000;64(3):139-44.
- 55. Fenaux P, Chastang C, Chevret S, Sanz M, Dombret H, Archimbaud E, et al. A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1999;94(4):1192-200.
- 56. Avvisati G, Lo CF, Diverio D, Falda M, Ferrara F, Lazzarino M, et al. AIDA (all-trans retinoic acid + idarubicin) in newly diagnosed acute promyelocytic leukemia: a Gruppo Italiano Malattie Ematologiche Maligne dell'Adulto (GIMEMA) pilot study. Blood. 1996;88(4):1390-8.
- 57. Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Woods WG, et al. All-trans retinoic acid in acute promyelocytic leukemia: long-term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood. 2002;100(13):4298-302.
- 58. Vahdat L, Maslak P, Miller WH, Jr., Eardley A, Heller G, Scheinberg DA, et al. Early mortality and the retinoic acid syndrome in acute promyelocytic leukemia: impact of leukocytosis, low-dose chemotherapy, PMN/RAR-alpha isoform, and CD13 expression in patients treated with all-trans retinoic acid. Blood. 1994;84(11):3843-9.
- 59. Larson RS, Tallman MS. Retinoic acid syndrome: manifestations, pathogenesis, and treatment. Best Pract Res Clin Haematol. 2003;16(3):453-61.
- 60. de BS, Dombret H, Sanz M, Miguel JS, Caillot D, Zittoun R, et al. Incidence, clinical features, and outcome of all trans-retinoic acid syndrome in 413 cases of newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1998;92(8):2712-8.
- 61. Breccia M, Diverio D, Noguera NI, Visani G, Santoro A, Locatelli F, et al. Clinico-biological features and outcome of acute promyelocytic leukemia patients with persistent polymerase chain reaction-detectable disease after the AIDA front-line induction and consolidation therapy. Haematologica. 2004;89(1):29-33.
- 62. Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 1996;88(3):1052-61.
- 63. Douer D, Tallman MS. Arsenic trioxide: new clinical experience with an old medication in hematologic malignancies. J Clin Oncol. 2005;23(10):2396-410.
- 64. Takeshita A, Shinjo K, Naito K, Matsui H, Sahara N, Shigeno K, et al. Efficacy of gemtuzumab ozogamicin on ATRA- and arsenic-resistant acute promyelocytic leukemia (APL) cells. Leukemia. 2005;19(8):1306-11.
- 65. Jácomo RH, Melo RAM, Souto FR, Mattos ER, Oliveira CO, Fagundes EF, et al. Clinical features and outcomes of 134 Brazilians with acute promyelocytic leukemia who received ATRA and anthracyclines. Haematologica. 2007;(92):1431-32.
- 66. Estey E, Garcia-Manero G, Ferrajoli A, Faderl S, Verstovsek S, Jones D, et al. Use of all-trans retinoic acid plus arsenic trioxide as an alternative to chemotherapy in untreated acute promyelocytic leukemia. Blood. 2006;107(9):3469-73.
Datas de Publicação
-
Publicação nesta coleção
01 Abr 2008 -
Data do Fascículo
Fev 2008
Histórico
-
Aceito
06 Nov 2007 -
Recebido
25 Jul 2007