Resumos
As hemofilias são doenças hemorrágicas resultantes da deficiência de fator VIII (hemofilia A) ou de fator IX (hemofilia B) da coagulação, decorrentes de mutações nos genes que codificam os fatores VIII ou IX, respectivamente. A hemofilia A é mais frequente que a hemofilia B e acomete aproximadamente 1:10.000 nascimentos masculinos. A gravidade e frequência dos episódios hemorrágicos está relacionado ao nível residual de atividade de fator VIII presente no plasma e este relaciona-se ao tipo de mutação associada à doença. A clonagem do gene do fator VIII tornou possível o conhecimento das bases moleculares da hemofilia A, sendo hoje conhecidas mais de 1.000 mutações associadas à doença. O conhecimento das bases moleculares da hemofilia A permite uma melhor compreensão da relação genótipo-fenótipo da doença, tomada de condutas clínicas diferenciadas em casos de mutações associadas a um maior risco de desenvolvimento de inibidor, determinação da condição de portadora de hemofilia em mulheres relacionadas aos pacientes, implementação de programa de aconselhamento genético/orientação familiar e melhor compreensão das relações estruturais-funcionais do gene-proteína. Este artigo propõe revisar as bases moleculares da hemofilia A, os métodos laboratoriais utilizados para a caracterização das mutações e as implicações clínicas envolvidas no diagnóstico molecular da hemofilia A.
Hemofilia A; Mutação; Diagnóstico molecular; Determinação de portadoras; Genes; Fator VIII
Hemophilias are bleeding disorders due to deficiency of the blood coagulation factor VIII (hemophilia A) or factor IX (hemophilia B), resulting from mutation on the gene coding for factor VIII or factor IX. Hemophilia A is more frequent than hemophilia B and affects 1:10,000 male newborns. The severity and frequency of hemorrhagic episodes is related to residual activity of factor VIII present in the plasma and relates to the type of mutation associated with the disorder. Cloning of the factor VIII gene has enabled researchers to better understand the molecular basis of hemophilia A, accounting to date, for more than 1,000 mutations associated with the disease. This comprehensive knowledge permits an improved comprehension of the genotype-phenotype relation, establishment of clinical policies when mutations related to higher risk of inhibitors development are known, identification of hemophilia carriers in case of women related to patients, implementation of a program of genetic counseling and discovery of structural-functional relationship between gene-protein. This article aims to review the molecular basis of hemophilia A, laboratory techniques used to characterize mutations and clinical implications involved in the molecular diagnosis of hemophilia A.
Hemophilia A; Mutation; Molecular diagnosis; Carrier detection; Genes; Factor VIII
ARTIGO DE REVISÃO
As bases moleculares da hemofilia A
Simone Ferreira PioI; Guilherme Corrêa de OliveiraII; Suely Meireles RezendeIII
IDoutoranda, Centro de Pesquisas Renè Rachou, FIOCRUZ. Fundação Hospitalar do Estado de Minas Gerais. Belo Horizonte, MG.
IIPesquisador, Centro de Pesquisas Renè Rachou, FIOCRUZ. Belo Horizonte, MG, Brasil
IIIProfessora-adjunta do Departamento de Clínica Médica da Faculdade de Medicina da Universidade Federal de Minas Gerais. Belo Horizonte, MG
RESUMO
As hemofilias são doenças hemorrágicas resultantes da deficiência de fator VIII (hemofilia A) ou de fator IX (hemofilia B) da coagulação, decorrentes de mutações nos genes que codificam os fatores VIII ou IX, respectivamente. A hemofilia A é mais frequente que a hemofilia B e acomete aproximadamente 1:10.000 nascimentos masculinos. A gravidade e frequência dos episódios hemorrágicos está relacionado ao nível residual de atividade de fator VIII presente no plasma e este relaciona-se ao tipo de mutação associada à doença. A clonagem do gene do fator VIII tornou possível o conhecimento das bases moleculares da hemofilia A, sendo hoje conhecidas mais de 1.000 mutações associadas à doença. O conhecimento das bases moleculares da hemofilia A permite uma melhor compreensão da relação genótipo-fenótipo da doença, tomada de condutas clínicas diferenciadas em casos de mutações associadas a um maior risco de desenvolvimento de inibidor, determinação da condição de portadora de hemofilia em mulheres relacionadas aos pacientes, implementação de programa de aconselhamento genético/orientação familiar e melhor compreensão das relações estruturais-funcionais do gene-proteína. Este artigo propõe revisar as bases moleculares da hemofilia A, os métodos laboratoriais utilizados para a caracterização das mutações e as implicações clínicas envolvidas no diagnóstico molecular da hemofilia A.
Unitermos: Hemofilia A. Mutação. Diagnóstico molecular. Determinação de portadoras. Genes. Fator VIII.
SUMMARY
Hemophilias are bleeding disorders due to deficiency of the blood coagulation factor VIII (hemophilia A) or factor IX (hemophilia B), resulting from mutation on the gene coding for factor VIII or factor IX. Hemophilia A is more frequent than hemophilia B and affects 1:10,000 male newborns. The severity and frequency of hemorrhagic episodes is related to residual activity of factor VIII present in the plasma and relates to the type of mutation associated with the disorder. Cloning of the factor VIII gene has enabled researchers to better understand the molecular basis of hemophilia A, accounting to date, for more than 1,000 mutations associated with the disease. This comprehensive knowledge permits an improved comprehension of the genotype-phenotype relation, establishment of clinical policies when mutations related to higher risk of inhibitors development are known, identification of hemophilia carriers in case of women related to patients, implementation of a program of genetic counseling and discovery of structural-functional relationship between gene-protein. This article aims to review the molecular basis of hemophilia A, laboratory techniques used to characterize mutations and clinical implications involved in the molecular diagnosis of hemophilia A.
Key words:Hemophilia A. Mutation. Molecular diagnosis. Carrier detection. Genes. Factor VIII.
INTRODUÇÃO
A coagulação do sangue consiste em uma série de reações bioquímicas sequenciais envolvendo a interação de proteínas, comumente referidas como fatores da coagulação, além de células (em particular, as plaquetas) e íons. O processo, em última análise, leva à formação de um coágulo, cujo principal componente é a fibrina1. Deficiências dos fatores da coagulação podem ocasionar doenças hemorrágicas (coagulopatias). As principais manifestações clínicas destas doenças são os sangramentos que podem ocorrer de forma espontânea ou induzida por trauma ou cirurgia. Dentre todas coagulopatias, as hemofilias e a doença de von Willebrand são as mais frequentes. O primeiro relato conhecido sobre hemofilia encontra-se em um decreto do Talmude (livro de condutas do povo judeu) datado do século II d.C. Neste decreto, o relator isenta da circuncisão crianças provenientes de famílias que tenham sofrido duas mortes decorrentes de hemorragias após o procedimento. Desde então, a hemofilia soma mais de 1.800 anos de história, contada por esparsos casos durante a Idade Média, por estudos pioneiros durante o século XIX e por avanços da bioquímica e genética ao longo do século XX2,3.
Aspectos gerais da hemofilia
Sabe-se hoje que as hemofilias são doenças resultantes da deficiência quantitativa dos fatores VIII (hemofilia A) ou IX (hemofilia B) da coagulação, podendo decorrer de fatores adquiridos ou hereditários. As formas adquiridas, mais raras, são resultantes do desenvolvimento de autoanticorpos, associados a doenças autoimunes, câncer ou causas de origem idiopática. A hemofilia hereditária resulta de alterações genéticas nos genes que codificam o fator VIII (hemofilia A) ou IX (hemofilia B) da coagulação4. A hemofilia A hereditária é o assunto desta revisão.
A hemofilia A, também conhecida como hemofilia clássica, é mais comum do que a hemofilia B na proporção de aproximadamente 4:1. Em diversas populações étnicas e geograficamente distintas já estudadas, a prevalência da hemofilia A é de 1:10.000 nascimentos masculinos. Esta frequência parece não variar consideravelmente entre as populações e tem sido mantida por um equilíbrio entre a perda de mutações, por exemplo, no caso de um paciente com hemofilia que tem somente filhos homens e não transmite aos mesmos a doença, e o aparecimento de mutações espontâneas5.
O quadro clínico da hemofilia A é marcado pela recorrência de hemorragias, principalmente em articulações (hemartroses) e músculos (hematomas), podendo também ocorrer em outros locais do corpo e, em casos mais graves, hemorragias internas e do sistema nervoso central. A frequência e a gravidade dos episódios hemorrágicos dependem do nível residual do fator VIII (FVIII:Ag) presente no plasma e sua respectiva atividade funcional (FVIII:C). É em função deste nível de concentração e de atividade que a hemofilia A é usualmente classificada nas formas grave (< 0,01 UI/mL de FVIII:Ag e/ou < 1% de FVIII:C), moderada (< 0,01- 0,05 UI/mL de FVIII:Ag e/ou < 1-5% de FVIII:C) e leve (> 0,05 a < 0,40 UI/mL de FVIII:Ag e/ou > 5% e < 40% de FVIII:C). A frequência de hemofilia leve, moderada e grave é de 50%, 10% e 40%, respectivamente6,7.
Aspectos genéticos da hemofilia
Por ser uma doença genética recessiva ligada ao cromosomo X, a hemofilia A afeta quase exclusivamente homens. Homens possuem um único alelo de fator VIII (XY) enquanto as mulheres possuem dois alelos (XX). Homens com um alelo com mutação (XHY, hemizigose) terão a doença, enquanto mulheres com um único alelo com mutação (XHX, heterozigose) serão portadoras e, portanto, com 50% de probabilidade de transmitir o alelo anormal à sua prole, em cada gestação. Já as mulheres com mutações em ambos os alelos (XHXH, homozigose) manifestarão a doença, embora esta seja uma situação muito rara (Figura 1A). Cerca de 30% dos pacientes diagnosticados com hemofilia A não possuem história familiar prévia da doença (Figura 1 B). Estes casos são explicados pela ocorrência de mutações espontâneas que podem acontecer em duas situações: (I) durante a gametogênese de um dos progenitores. Neste caso, a mutação ocorre em uma das células da linhagem germinativa e persiste em todos os descendentes clonais da mesma e em uma proporção dos gametas, caracterizando o mosaicismo germinativo; (II) a mutação pode também ocorrer durante os primeiros estágios da embriogênese do paciente afetado. Nesta situação, uma das células, presentes nos estágios iniciais do embrião em formação, sofre mutação e esta é repassada às demais células por meio de mitoses sucessivas, fazendo com que, o indivíduo possua tanto células normais quanto alteradas. Este fato caracteriza o mosaicismo somático8,9.

O gene do fator VIII
Enquanto aspectos clínicos e bioquímicos da hemofilia A são conhecidos há décadas, as bases moleculares da doença somente foram compreendidas a partir de 1984 após a clonagem e caracterização do gene que codifica o fator VIII (F8). Este gene está localizado na extremidade do braço longo do cromossomo X (porção Xq28) e compreende 186.000 pb (pares de bases) distribuídos entre 26 éxons e 25 íntrons que variam de 69 a 3.106 pb e de 0,2 a 32,4 kb (kilobases, 1.000 pares de bases), respectivamente. Toda a sequência do gene compreende aproximadamente 9 kb de éxons e 177 kb de íntrons que, juntos, correspondem a cerca de 0,1% de todo o cromossomo X10 (Figura 2). Outras características de F8 devem ainda ser enfatizadas: (I) a presença de cerca de 70 ilhas CpG, das quais 2% estão em sequências codificadoras; (II) o grande tamanho do éxon 14 (3.106 pb), que representa cerca de 40% da região codificadora, mas que codifica a parte funcionalmente menos importante da proteína (Figura 2) e (III) o grande tamanho do íntron 22, que apresenta uma ilha de CpG que funciona como promotor bidirecional para dois transcritos adicionais denominados F8A e F8B11

F8A possui 1,8 kb, não apresenta íntrons e é transcrito em direção oposta a F8. Já F8B possui 2,5 kb e é transcrito na mesma direção de F8, sendo parcialmente sobreposto ao mesmo (éxons 23 -26). As sequências de F8A e da ilha de CpG formam uma região de aproximadamente 9,5 kb denominadas int22h1. Esta sequência possui similaridade com duas cópias extragênicas (int22h2 e int22h3) localizadas aproximadamente entre 400-500 kb anteriores a porção telomérica de F8. A ocorrência de uma segunda região com alto grau de similaridade envolvendo sequência intra e extragênica pode ser observada neste gene. Esta corresponde a uma região de aproximadamente 1 kb, mais exatamente 1.041 pb, presente no íntron 1, denominada int1h1. A região de alta similaridade correspondente está localizada a aproximadamente 140 kb anteriores a porção telomérica de F8. Essa cópia extragênica é denominada int1h2. Estas regiões de alta similaridade funcionam como sítios de recombinação homóloga e determinam rearranjos intragênicos12,13 (Figura 3).
Outra característica importante deste gene é seu baixo índice de polimorfismo. Os polimorfismos observados neste gene são de dois tipos: SNPs (polimorfismo de único nucleotídeo) e VNTRs (sequências de número variável repetidas em "tandem"). Essas variações têm relevância clínica no contexto de doenças hereditárias, uma vez que podem ser usados para rastrear um gene mutado em uma família afetada. Entretanto, deve-se considerar as diferenças nas frequências de alelos entre populações distintas, pois loci de grande interesse diagnóstico podem diferir de acordo com a etnia da população, assim como com o desequilíbrio de ligação14.
Do mRNA do fator VIII à proteína
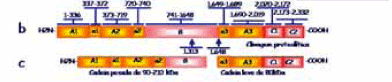
O transcrito de F8 tem cerca de 9010 b de tamanho com uma curta região 5' não traduzida (150 pb), uma janela de leitura aberta (7.056 bases) e uma longa extremidade 3'(1.806 pb) não traduzida. A janela de leitura aberta codifica uma proteína precursora de cadeia única contendo 2.351 aminoácidos, 19 destes correspondentes à sequência de um peptídeo sinal. Durante a passagem da proteína pelo retículo endoplasmático, o peptídeo sinal é clivado resultando em uma proteína de estrutura primária de 2.332 aminoácidos15. A análise da estrutura primária desta proteína mostrou a presença de domínios: A1-a1-A2-a2-B-a3-A3-C1-C2 (Figura 2:4b). O conhecimento da via de síntese intracelular do fator VIII tornou-se possível graças aos estudos com cultura de linhagens de células de mamíferos, não humanas, transfectadas com o cDNA do fator VIII. A maturação e a secreção do fator recém sintetizado envolvem grande variedade de interações proteína-proteína e diversas modificações pós-traducionais. No retículo endoplasmático, o fator VIII sofre vários eventos de glicosilação para então ser encaminhado para o complexo de Golgi. Neste, a molécula sofrerá processo de sulfatação e glicosilação de resíduos específicos, além de clivagens proteolíticas na junção dos domínios B-A3 e em sítios adicionais dentro do domínio "B". Estas clivagens dividem a molécula de fator VIII em duas porções: (I) cadeia pesada, constituída pelo domínio A1, A2 e fragmentos heterogêneos do domínio B e (II) cadeia leve, constituída pelos domínios A3, C1 e C2, resultando na secreção de um heterodímero dependente de metais bivalentes16 (Figura 2:4d).
O fator VIII no plasma
Imediatamente após a secreção do fator VIII, o heterodímero interage com o fator de von Willebrand que atua como carreador do fator VIII durante sua circulação no plasma. O fator VIII, ligado ao fator de von Willebrand, terá uma meia vida que varia de 12-15 horas. O fator VIII é ativado pela trombina e pelo fator X. Ambas as proteases clivam a molécula de fator VIII nas posições Arg372 e Arg740 da cadeia pesada e Arg1689 da cadeia leve (Figura 2:4e). Essas clivagens resultam em dois fragmentos A1-A2 e A3-C1-C2 que permanecem associados por meio de interações eletrostáticas entre o domínio A1 e A3. A ativação do fator VIII reduz drasticamente a afinidade com o fator de von Willebrand. O fator VIII ativado juntamente com fator IX ativado, fosfolípides de membrana carregados negativamente e íons cálcio formam o complexo tenase, que por sua vez, atua enzimaticamente sobre o fator X convertendo-o em sua forma ativada. O fator X ativado participa da conversão da protrombina em trombina juntamente com fator V ativado e íons cálcio (complexo protrombinase). A trombina é a enzima chave da cascata de coagulação que, em última instância, converterá fibrinogênio em fibrina. Uma vez que o fator VIII ativado tenha desempenhado seu papel na cascata da coagulação, este é rapidamente inativado. O mecanismo exato desta inativação sob condições fisiológicas ainda é desconhecido. Entretanto, a proteína C ativada cliva a cadeia pesada do fator VIII nas posições Arg336/Met337 e Arg562/Gly563 em uma reação que requer a presença de proteína S e a região carboxi-terminal do domínio B do fator V17-19.
A heterogeneidade das mutações na hemofilia A
Desde a clonagem de F8 em 1984, as bases moleculares da hemofilia A vem sendo extensivamente estudadas. Estes estudos geraram grande volume de informações que hoje são compiladas em bancos de dados acessíveis através da internet. Um dos mais representativos bancos de informação sobre hemofilia A é o HAMSTeRS (Haemophilia AMutation Search Test and Resource Site).20 Este banco disponibiliza a listagem completa das mutações publicadas, dados fenotípicos, modelos moleculares, métodos para o rastreamento de mutações, lista de polimorfismos e uma revisão sobre a patologia molecular da hemofilia A. O HAMSTeRS lista atualmente mais de 1.000 mutações associadas à hemofilia A envolvendo substituições, deleções e inserções distribuídas por todo o gene21. Os dados moleculares sobre a hemofilia A demonstram uma grande heterogeneidade de mutações associadas à doença. Entretanto, duas mutações específicas, a inversão do íntron 22 e a inversão do íntron 1, são encontradas em cerca de 50% e 5% dos casos de hemofilia A grave, respectivamente22.
A inversão do intron 22 resulta da recombinação homóloga entre a sequência int22h1 e uma das cópias extragênicas (int22h2 ou int22h3) localizadas a 400-500 kb anteriores à porção telomérica de F8. Dependendo da cópia extragênica envolvida no evento de recombinação, dois tipos principais de inversão são reconhecidos. A inversão do tipo 1 envolve a cópia distal (int22h3) e a inversão do tipo 2 envolve a cópia proximal (int22h2). Estas inversões ocorrem quase exclusivamente na meiose masculina23,24 (Figura 3).
A inversão do íntron 1 envolve a recombinação homóloga entre uma região de 1.041 pb situada no íntron 1 (int1h1) e uma cópia extragênica (int1h2) situada a aproximadamente a 140 kb anterior da porção telomérica de F8 entre os genes C61A e VBP1. A similaridade entre as sequências int1h1 e int1h2 é muito alta, sendo observada a diferença de um único nucleotídeo entre elas25,26 (Figura 3).
Apesar do grande volume de informações sobre as bases moleculares da hemofilia A, existe um percentual de cerca de 2% dos pacientes cujo diagnóstico molecular não é estabelecido, mesmo usando as abordagens mais avançadas para a análise do gene. Isto pode ser decorrente de mutações que estão presentes em regiões não codificadoras ou que estes pacientes não possuem mutação associada ao F8. É possível que haja participação de outros genes, por exemplo, no caso dos genes LMAN1 (do inglês, lectin, manose binding 1, também conhecida como ERGIC53) e MCFD2 (do inglês, multiple coagulation factor deficiency 2 protein) responsáveis pelo transporte intracelular do fator VIII do retículo endoplasmático para o aparato de Golgi. Defeitos destas proteínas resultam em deficiência combinada de fatores V e VIII da coagulação27.
Diversas proteínas interagem com o fator VIII na circulação. Uma das mais importantes é o fator de von Willebrand. O fator de von Willebrand é uma glicoproteína multimérica que desempenha dois principais papéis na hemostasia: (I) ela participa da hemostasia primária quando atua como um mediador para a interação entre as glicoproteinas da superfície plaquetária e a parede do endotélio, no sítio de injúria vascular e, (II) participa da hemostasia secundária quando atua como proteína carreadora do fator VIII, aumentando a meia vida deste.28 Considerando seu papel na hemostasia secundária, é fácil compreender que a deficiência de fator de von Willebrand resulta em uma correspondente diminuição dos níveis de fator VIII e em quadros clínicos similares àqueles observados em pacientes com hemofilia A. Dos diversos subtipos clínicos da doença de von Willebrand (tipos 1, 2A, 2B, 2M, 2N), a variante 2N é a única que decorre da redução da interação entre fator von Willebrand e fator VIII devido a mutações nos domínios D' ou D3 daquele fator29. Homozigotos ou heterozigotos compostos, para essa condição, apresentam redução dos níveis de fator VIII no plasma e, geralmente, apresentam eventos hemorrágicos, que muitas vezes são erroneamente diagnosticados como hemofilia A. A análise desta variante é importante, sobretudo em pacientes cuja análise molecular para hemofilia A mostrou-se negativa para mutações em F830. Outra questão a ser pontuada quanto a mutações em outros genes diz respeito a fatores que podem influenciar o fenótipo da hemofilia. Trabalhos recentes mostraram que, mesmo pacientes que apresentam a mesma mutação e com níveis residuais de fator VIII similares, podem apresentar variações quanto ao padrão de manifestações hemorrágicas. Dentre as explicações para a variabilidade fenotípica destaca-se a presença das mutações protrombóticas, tais como o fator V Leiden, mutações no gene da protrombina, (G20210A), no gene da metilenotetrahidrofolato redutase, deficiência de proteína C e proteína S, ou mesmo co-herança de polimorfismos que afetam o sistema de coagulação. Há ainda trabalhos recentes que associam a variabilidade fenotípica a diferenças da farmacocinética do fator VIII infundido nos pacientes. Estes fatores podem minimizar a gravidade clínica da hemofilia A31,32.
O diagnóstico molecular da hemofilia A
A história familiar e a ocorrência de episódios hemorrágicos são os principais dados para o diagnóstico da hemofilia. Entretanto, cerca de 20%-30% dos pacientes com hemofilia não apresentam história familiar da doença. A disponibilidade de técnicas moleculares acrescentou, ao diagnóstico laboratorial da hemofilia, a possibilidade de caracterizar a doença em nível molecular. A análise molecular pode utilizar tanto DNA ou RNA, embora o primeiro seja mais utilizado em função de sua maior estabilidade e facilidade de manuseio. As abordagens utilizadas para a análise do DNA podem ser diretas ou indiretas. As abordagens indiretas são baseadas em análise de ligação e empregam marcadores polimórficos específicos. As abordagens diretas são baseadas em diversas técnicas moleculares que permitem a identificação precisa da mutação. Entretanto, apresentam como dificuldade o extenso tamanho de F8, a complexidade genômica do mesmo e a grande diversidade de mutações. Mutações envolvendo grandes sequências de DNA, isto é, grandes deleções, inserções ou rearranjos podem ser identificadas por meio de análise de Southern blotting 33. Para a detecção da inversão do intron 22, vêm sendo utilizadas a técnicas de PCR longa propostas por Liu et al.34, e, mais recentemente, uma técnica de PCR multiplex proposta por Rossetti et al.35 Mutações envolvendo pequenas alterações na sequência de DNA (em geral, mutações em ponto) requerem procedimentos técnicos especiais. Para tal, utiliza-se a amplificação por PCR, seguida de um ensaio de mobilidade eletroforética (DGGE - Denaturing, Gradient Gel eletrophoresis, SSCP - Single Strand Conformation Polymorphism, CSGE - Conformation Sensitive Gel Eletrophoresis, DHPLC - Denaturing High Performance Liquid Chromatography, e CCMA - Chemical Cleavage Mismatch Analysis) e/ou de sequenciamento36-37. De modo geral, as taxas de detecção de mutações obtidas com estes ensaios são bastante elevadas e têm sido aperfeiçoadas mediante adequação de protocolos.
Implicações clínicas do diagnóstico molecular das hemofilias
Em geral, a caracterização molecular da hemofilia A não é necessária para o diagnóstico da doença, nem para o seu tratamento. O diagnóstico da hemofilia é suspeitado clinicamente e confirmado através da realização do teste de FVIII:C. O tratamento é realizado através de infusão do fator deficiente de forma profilática ou sob demanda. Entretanto, o diagnóstico molecular das hemofilias é altamente relevante para a identificação de portadoras, naquelas situações em que a condição é possível, mas não obrigatória (filhas de mulher portadora) ou, ainda, naqueles casos em que, embora não exista história familiar prévia de hemofilia, ocorreu nascimento de indivíduo com a doença. Nestas situações, a identificação da condição de portadora é fundamental, particularmente em mulheres em idade fértil, pois permite que a mulher possa participar e decidir sobre o planejamento de sua família. Enfim, o diagnóstico molecular das hemofilias permite estruturar um programa de aconselhamento genético/orientação familiar que permite determinar com precisão a condição genética da doença (hereditária ou esporádica). Os dados advindos dessa determinação disponibilizam potenciais informações quanto às características da doença, riscos de recorrência, modalidades de transmissão genética, arsenal diagnóstico pré e pós-natal assim como suporte psicológico e emocional38,39.
O diagnóstico molecular das hemofilias tem importância prática no diagnóstico pré-natal em países onde o abortamento de indivíduos afetados por doenças graves é legal. Esta situação não é permitida no Brasil, tornando seu uso limitado para esta indicação. Uma das complicações mais temíveis da hemofilia A é o desenvolvimento de inibidores de fator VIII (aloanticorpos), que ocorre em 10%-30% dos pacientes. Pacientes com inibidor tornam-se refratários a infusão de fator VIII, apresentam hemorragias mais frequentes e/ou mais graves e necessitam tratamento com produtos mais onerosos. Em aproximadamente 95% dos casos, o desenvolvimento de inibidor ocorre nos primeiros 50 dias de infusão do concentrado. O único tratamento capaz de erradicar inibidores em hemofilia é a imunotolerância, que consiste na infusão frequente de concentrado de fator VIII, com fins de "tolerizar" o paciente. Estudos correlacionando o desenvolvimento de inibidores e os tipos de mutação demonstraram que mutações com troca de aminoácidos e pequenas deleções exibiram baixa prevalência de inibidores (cerca de 5%). Em contraste, grandes deleções, mutações sem sentido e a prevalente inversão do íntron 22 se associaram a maior prevalência (7-10 vezes) de inibidores (cerca de 35%).40 Resultados semelhantes foram demonstrados por Goodeve e Peake41 ao estabelecerem a correlação entre genótipo e fenótipo e o desenvolvimento de inibidores. É possível que pacientes com mutações de troca de sentido sejam capazes de sintetizar fator VIII endógeno, que, embora não funcional, seria suficiente para induzir a imunotolerância para o fator VIII exógeno. Em contraste, quantidades mínimas de fator VIII endógeno são sintetizadas em pacientes com defeitos moleculares graves e o fator VIII exógeno representaria, portanto, uma proteína estranha que desencadearia uma resposta imune 40. Ainda, mutações na cadeia leve do fator VIII parecem conferir maior risco de inibidores do que mutações na cadeia pesada 42. A maioria dos inibidores em pacientes com hemofilia A leve/moderada ocorre em associação com mutações com sentido trocado nos domínios A1-A2 (entre os resíduos 482-501) e na junção C1-C2 do fator VIII. Estas mutações estão associadas a risco aumentado de inibidor em até 50%, possivelmente ocasionado por mudança conformacional da proteína 43,44. Em particular, algumas mutações se encontram mais comumente associadas ao desenvolvimento de inibidor em hemofilia A leve/moderada: Arg593Cys, Arg2150His e Trp2229Cys.45 Em pacientes com hemofilia A leve/moderada e mutações de risco para desenvolvimento de inibidores, recomenda-se tratar os eventos hemorrágicos com desmopressina sempre que possível a fim de minimizar as infusões de concentrado de fator VIII, reduzindo, assim, o risco de desenvolvimento de inibidor. Ainda, diante de mutações de risco, recomenda-se testar o paciente para a presença do inibidor mais frequentemente, particularmente até a 50ª infusão. Um estudo recente revelou que pacientes com hemofilia A e inibidor, tratados com imunotolerância, que apresentam grandes deleções de F8, apresentam resposta pior em comparação com outros tipos de mutação 46. O estudo de mutações naturais pode levar a uma compreensão mais clara das relações estrutural-funcionais de diversas proteínas. Este aprendizado não é diferente com relação ao fator VIII, uma vez que a caracterização de diversas mutações tem ocasionado uma melhor compreensão das relações F8-fator VIII47,48.
Por último, o diagnóstico molecular da hemofilia A é fundamental para o aconselhamento genético. Especialmente em casos esporádicos, sem história familiar prévia da doença, somente o conhecimento da mutação, associada pode reconhecer a condição de portadora e predizer com precisão o risco de recorrência da doença na futura prole. Ainda, o conhecimento do genótipo pode trazer vantagens na predição do risco de desenvolvimento de inibidores, possivelmente do sucesso da imunotolerância e, no futuro, para terapia gênica. Recomenda-se que o diagnóstico molecular da hemofilia A seja realizado em todos os pacientes acometidos pela doença. Em países desenvolvidos, o teste é ofertado a todos os pacientes com hemofilia ao diagnóstico, embora não seja ainda de rotina no Brasil.
FINANCIAMENTO
Fundação de Amparo À Pesquisa do Estado de Minas (FAPEMIG)
Artigo recebido: 24/03/08
Aceito para publicação: 01/07/08
Trabalho realizado pelo Centro de Pesquisas Renè Rachou, FIOCRUZ; Fundação Hospitalar do Estado de Minas Gerais; Universidade Federal de Minas Gerais, Belo Horizonte, MG
* Correspondência: Departamento de Clínica Médica - Faculdade de Medicina. Avenida Alfredo Balena, 190 - 2º andar - sala 243. CEP 30130-110 - Belo Horizonte - MG. Tel/FAX: (31) 3409-9746/45. srezende@medicina.ufmg.br
- 1. Hirsh J, Weitz JI. Thrombosis and anticoagulation. Semin Hematol. 1999;(4 Suppl 7):118-32.
- 2. DiMichele D, Neufeld EJ. Hemophilia. A new approach to an old disease. Hematol Oncol Clin North Am. 1998;12(6): 1315-1344.
- 3. Mannucci PM, Tuddenbam EG. The hemophilias: progress and problems. Semin Hematol. 1999;36(4 Suppl 7):104-17.
- 4. Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003; 361:1801-1809.
- 5. Hedner U, Ginsburg D, Lusher JM, High KA. Congenital hemorrhagic disorders: new insights into the pathophysiology and treatment of hemophilia. Hematology Am Soc Hematol Educ Program. 2000;241-65.
- 6. White GC, Rosendaal F, Aledor, LM, Lusher JM, Rothschild C, Ingerslev J. International Society on Thrombosis and Haemostasis: Subcommitee on factor VIII and Factor IX of the Scientific and Standardization; 2000.
- 7. Graw J, Brackmann H, Oldenburg J, Scheneppeheim R, Spannag M, Schwaab, R. Nat Rev Genet. 2005;6:488-501.
- 8. Cohen AJ, Kessler CM. Treatment of inherited coagulation disorders. Am J Med. 1995;99:675-682.
- 9. Soares RPS, Chamone DAF, Bydlowski SP. Factor VIII gene inversion and polymorphism in Brazilian patients with haemophilia A: carrier detection and prenatal diagnosis. Haemophilia. 2001;7:299-305.
- 10. Gitshier J, Wood WI, Goralka, TM, Wion, KL, Chen, EY, Eaton DH, et al. Characterization of the human factor VIII gene. Nature. 1984;312:326-30.
- 11. Oldenburg J,Brackmann H, Hanfland P, Schwaab R. Molecular genetics in hemophilia A. Vox Sang. 2000;78(Suppl 2):33-8.
- 12. Brinke A, Tagliavacca L, Naylor J, Green PG, Giannelli F. Two chimaeric transcription units result from an inversion breaking intron 1 of the factor VIII gene and a region reportedly affected by reciprocal translocations in T-cel leukaemia. Hum Mol Genet. 1996;5:1945-51.
- 13. Antonarakis SE. Molecular genetics of coagulation factor VIII gene and haemophila A. Haemophilia. 1998;4(Suppl 2):1-11.
- 14. Bowen DJ. Haemophilia A and haemophilia B: molecular insights. Mol Pathol. 2002;55:127-44.
- 15. Wood WI, Capon DJ, Simonsen CC, Eaton DL, Gitschier J, Keyt, B, et al. Expression of active human factor VIII from recombinant DNA clones. Nature. 1984;312(5992):330-3.
- 16. Scheiflinger F, Dorner F. Recent advances in the understanding of the molecular biology of hemophilia A: possible implications toward a more effective therapeutic regime. Wien Klin Wochenschr. 1999;115:172-80.
- 17. Ananyeva NM, Kouiavskaia DV, Shima M, Saenko EL. Catabolism of the coagulation factor VIII: can we prolong lifetime of FVIII in circulation? Trends Cardiovasc Med. 2001;11:251-7.
- 18. Saenko EL, Ananyeva NM, Tuddenham EGD,Kemball-Cook G. Factor VIII- novel insights into form and function. Br J Haematol.2002;119:323-31.
- 19. Rezende SM, Simmonds R E,Lane D.A. Coagulation, inflammation, and apoptosis: different roles for protein S and the protein S-C4b binding protein complex. Blood. 2004;103:1192-201.
-
20HAMSTeR. Haemophilia A mutation search test and resource site. [cited 2008]. Available from: http://europium.mrc.rpms.ac.uk/
- 21. Kemball-Cook G, Tuddenham EGD, Wacey AI. The factor VIII Structure and Mutation Resource Site: HAMSTeRS version 4. Nucleic Acids Res. 1998;26(1):216-9.
- 22. Andrikovics H, Klein I, Bors A, Nemes L, Marosi A,Váradi A, Tordai A. Analysis of structural changes of the factor VIII gene, involving intron 1 and 22, in severe hemophilia A. Haematologica. 2003;88:778-84.
- 23. Lakich D, Kazazian HH Jr, Antonarakis SE,Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. 1993;5:236-41.
- 24. Rossinter JP, Young M, Kimberland ML,. Hutter P, Ketterling RP, Gitschier J, et al. Factor VIII gene inversion causing severe hemophilia A originate almost exclusively in male germ cells. Hum Mol Genet. 1994;7:1035-9.
- 25. Brinke A, Tagliavacca L, Naylor J, Green PG, Giannelli F. Two chimaeric transcription units result from an inversion breaking intron 1 of the factor VIII gene and a region reportedly affected by reciprocal translocations in T-cell leukaemia. Hum Mol Genet. 1996;12:1945-51.
- 26. Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99:168-74.
- 27. Zhang B, Ginsburg D. Familial multiple coagulation factor deficiencies: new biologic insight from rare genetic bleeding disorders. J Thromb Haemost. 2004;2:1564-72.
- 28. De Wit TR, Van Mourik JA. Biosynthesis, processing and secretion of von Willebrand factor: biological implications. Best Pract Res Clin Haematol. 2001;14:241-55.
- 29. Mazurier C, Hilbert L. Type 2N von Willebrand disease. Curr Hematol Rep. 2005;4:350-8.
- 30. Schneppenheim R, Budde U, Krey S, Drewke E, Bergmann F, Lechler E, Oldenburg J, Schwaab R. Results of a screening for von Willebrand disease type 2N in patients with suspected haemophilia A or von Willebrand disease type 1. Thromb Haemost. 1996;76:598-602.
- 31. Dargaud Y, Meunier S, Negrier C. Haemophilia and thrombophilia: an unexpected association! Haemophilia. 2004;10:319-26.
- 32. Van Dijk K, Van Der Bom JG, Lenting PJ, De Groot PG, Mauser-Bunschoten EP, Roosendaal G, et al. HM. Factor VIII half-life and clinical phenotype of severe hemophilia A.Haematologica.2005;90:494-8.
- 33. Saweck J, Skulimouiska J, Windyga J, Lopaciuk S, Koscielak J. Prevalence of the intron 22 inversion of the fator VIII gene and inhibitor development in polish patients with severe hemophilia A. Arch Immunol Ther Exp. 2005;53:352-6.
- 34. Liu Q, Nozari G, Sommer SS. Single-tube polymerase chain reaction for rapid diagnosis of the inversion hotspot of mutation in hemophilia A. Blood. 1998;92:1458-9.
- 35. Rossetti LC Radic CP, Larripa IB, De Brasi CD. Genotyping the hemophilia inversion hotspot by use of inverse PCR.Clin Chem. 2005;51:1154-8.
- 36. Goodeve AC. Laboratory methods for the genetic diagnostic of bleeding disorders. Clin Lab Haematol. 1998;20:3-19.
- 37. Vidal F, Farssac, E, Altisent C, Puig L, Gallardo D. Rapid hemophilia A molecular diagnosis by a simple DNA sequencing procedure: identification of 14 novel mutaton. Thromb Haemost. 2001;85:580-3.
- 38. Tagariello G, Belvoni D, Salviato R, Are A, De Biasi E., Goodeve A, et al. Experience of a single Italian center in genetic counseling for hemophilia: from linkage anayisis to molecular diagnosis. Haematologica. 2000;85:525-9.
- 39. Miller R. Cousenling about dianosis and inheritance og genetic bleeding disorders: Haemophilia A and B. Hemophilia. 1999;5:77-83.
- 40. Oldenburg J,Brackmann H, Schwaa, R. Risk factor for inhibitor development in hemophilia A. Haematologica. 2000;85(Suppl 10):7-17.
- 41. Goodeve A, Peake IR.The molecular basis hemophilia a: genotype-phenotype relationships and inhibitor development. Semin Thromb Hemost. 2003;29:23-30.
- 42. Goodeve A. The incidence of inhibitor development according to specific mutations and treatment. Blood Coagul Fibrinolysis. 2003;14 (Suppl 1):17-21.
- 43. Oldenburg J, Picard JK, Schwaab R, Brackmann HH, Tuddenham EG, Simpson E. HLA Genotype of patients with severe haemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost. 1997;77:238-42.
- 44. Hay CR. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia. 1998;4(4):558-63.
- 45. Hay CR, Ludlam CA, Colvin BT, Hill FG, Preston FE, Wasseem N, et al. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Thromb Haemost. 1998;79:762-6.
- 46. Salviato R, Belvini D, Radossi P, Sartori R, Pierobon F, Zanotto D, et al. F8 gene mutation profile and ITT response in a cohort of Italian haemophilia A patients with inhibitors. Haemophilia. 2007;13:361-72.
- 47. Spiegel PC, Murphy P, Stoddard BL. Surface-exposed hemophilic mutations across the factor VIII C2 domain have variable effects on stability and binding activities. J Biol Chem. 2004;279:53691-8.
- 48. Harbat D, Kalabova D, Novotny M, Vorlova Z. Thirty four novel mutation detected in factor VIII gene by multiplex CSGE: modeling of 13 novel aminoacid substitutions. J Thromb Haemost. 2003;1(4):773-81.
Datas de Publicação
-
Publicação nesta coleção
29 Maio 2009 -
Data do Fascículo
2009
Histórico
-
Recebido
24 Mar 2008 -
Aceito
01 Jul 2008