Summary
Retinoblastomas (RB) are the main forms of intraocular tumor in childhood, with a worldwide incidence of 1 case per 15,000 to 20,000 live births. Trilateral RB (RBT) is a rare combination of unilateral or bilateral RB with a midline intracranial neoplasm of neuroblastic origin, usually found in the pineal region or the suprasellar region, presenting variable incidence of 0.5% up to 6% among patients with RB. The article reports a case of unilateral RBT in a patient treated at Hospital A.C.Camargo.
Keywords:
retinoblastoma; eye neoplasms; infant
Resumo
O retinoblastoma (RB) é a principal forma de tumor intraocular na infância, apresentando uma incidência mundial de 1 caso em cada 15 mil a 20 mil nascidos vivos. O RB trilateral (RBT) é uma rara combinação de RB unilateral ou bilateral com uma neoplasia da linha média intracraniana de origem neuroblástica, geralmente na região da glândula pineal ou na região suprasselar, apresentando incidência variável de 0,5 a 6% entre pacientes com RB. O artigo relata o caso de uma paciente com RBT com acometimento unilateral atendida no Hospital A.C.Camargo.
Palavras-chave:
retinoblastoma; neoplasias oculares; lactente
Retinoblastomas (RB) are the main forms of intraocular tumor in childhood, with a worldwide incidence of 1 case per 15,000 to 20,000 live births.11 Kivela T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009; 93(9):1129-31.,22 Boubacar T, Fatou S, Fousseyni T, Mariam S, Fatoumata DT, Toumani S, et al. A 30-month prospective study on the treatment of retinoblastoma in the Gabriel Toure Teaching Hospital, Bamako, Mali. Br J Ophthalmol. 2010; 94(4):467-9. It is a tumor caused by inactivation of both copies of the Rb1 tumor suppressor gene, located on chromosome 13q14. The clinical presentation may be divided into hereditary (40% of cases) and somatic (60% of cases), according to two different mutational events. In the inherited form (also known as germinal) the first mutation occurs in the germ cell or is inherited from a parent, while the second occurs within the somatic cell (retinal). In these cases, the presentation is multicentric (bilateral or unilateral multifocal), the patient carries the defective gene in every cell of his or her body and can pass it on to his or her offspring. In the somatic form, both mutational events occur within the retinal cell, resulting in unilateral, unifocal, non-communicable disease.33 Knudson Jr AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971; 68(4):820-3.
Trilateral RB (RBT) is a rare combination of unilateral or bilateral RB with a midline intracranial neoplasm of neuroblastic origin (primary neuroectodermal tumor), usually found in the pineal region or the suprasellar region.44 Kivelä T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999; 17(6):1829-37. RBT can occur with both familial and sporadic forms of retinoblastoma with variable incidence of 0.5-2% in cases of a unilateral RB and up to 6% among the patients with bilateral RB.44 Kivelä T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999; 17(6):1829-37.,55 Shields CL, Meadows AT, Shields JA, Carvalho C, Smith AF. Chemoreduction for retinoblastoma may prevent intracranial neuroblastic malignancy (trilateral retinoblastoma). Arch Ophthalmol. 2001; 119(9):1269-72. The term trilateral retinoblastoma is used because the pineal is considered a photosensitive body (third eye) in lower vertebrates.66 Bader JL, Meadows AT, Zimmerman LE, Rorke LB, Voute PA, Champion LA, et al. Bilateral retinoblastoma with ectopic intracranial retinoblastoma: Trilateral retinoblastoma. Cancer Genet Cytogenet. 1982; 5(3):203-13.
The article reports the case of a patient aged 1 year and 4 months, who was initially examined at their town of origin (Maringá, state of Paraná, Brazil), after the mother perceived difference in the red reflex of the left eye compared to the right eye. She, then, sought evaluation by an ophthalmologist in the city of origin, being referred to the Hospital A.C.Camargo with suspected tumor in his left eye. After ophthalmologic evaluation, including retinal mapping and ocular ultrasound, the diagnosis of retinoblastoma was raised and screening tests requested for extra-ocular injury, including contrast-enhanced MRI of skull and orbits (MRI-SO). On MRI-SO, besides the intra-ocular lesion, another lesion in the pineal gland (Figure 1) was seen. The diagnosis made was RBT with monocular involvement. The patient was then subjected to 4 cycles of chemotherapy with Carboplatin and Etoposide in the period from 1/23/13 to 4/10/13. Enucleation of the left eyeball was performed on 03/6/13 (Figure 2). During patient’s progression, persistent pinealoblastoma was found, and radiotherapy (RDT) started with 25 Gy/5F on 9/13/13 until its completion, on 10/2/13. The patient’s condition progressed steadily in subsequent follow-ups, without signs of systemic disease or impairment of the right eye.
(A) MRI with contrast sagittal T1-weighted image. (B) MRI with contrast coronal T1-weighted image. (C) MRI with contrast axial T1-weighted image.
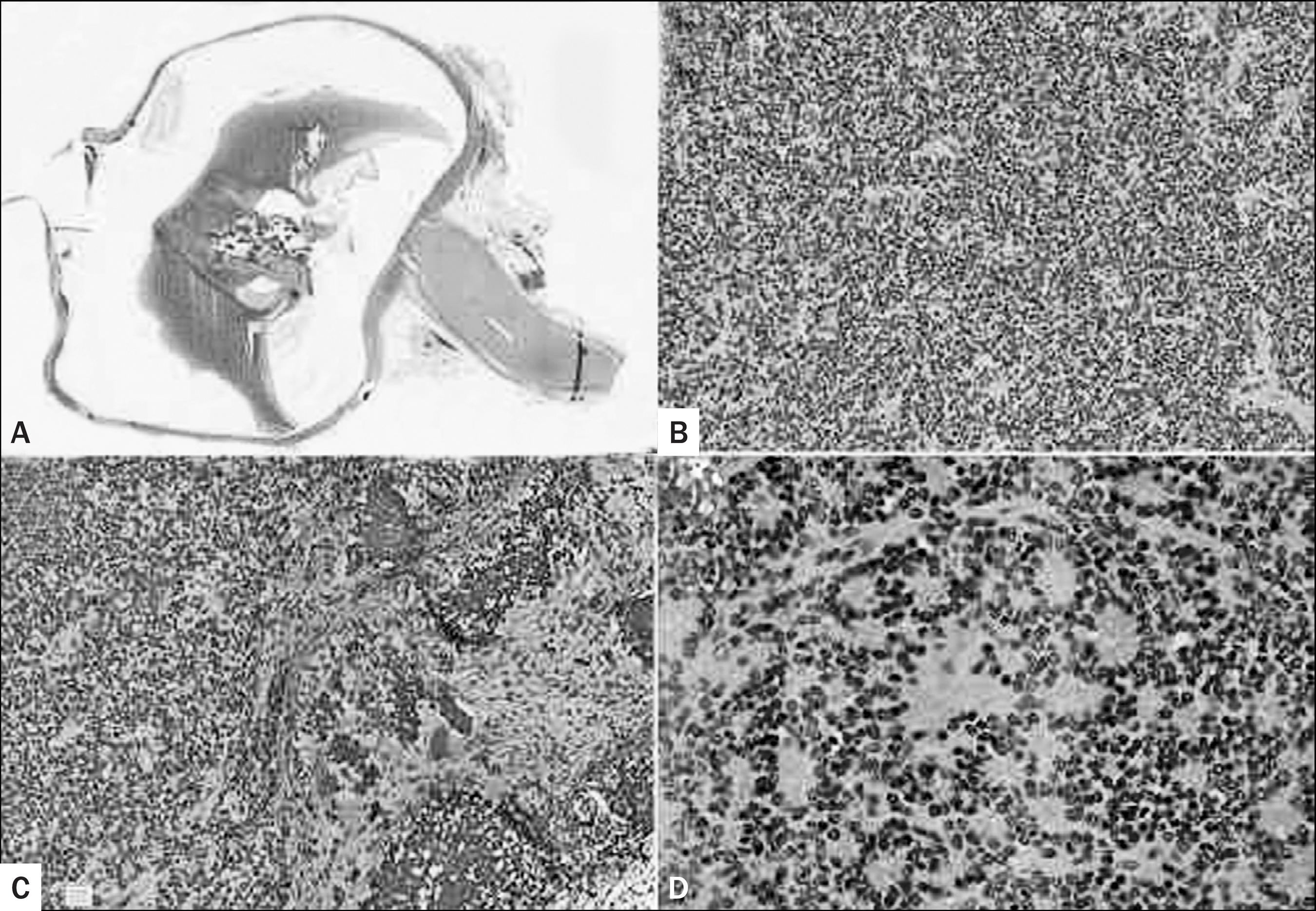
Retinoblastoma. (A) Histological section of left eyeball showing a 1.3 cm tumor in the posterior chamber, connected to the retina without choroidal or optic nerve infiltration (H&E, 0.5x). (B) Greater magnification showing a malignant neoplasm of small round blue cells, (C) with multiple foci of dystrophic calcifications (H&E, 20x). (D) Homer-Wright rosettes were focally present, but Flexner-Wintersteiner rosettes were not seen (H&E, 20x).
With early diagnosis, the survival rate for retinoblastoma is good, reaching 93% at 5 years. In cases of RBT, prognosis is worse with survival rate between 9 and 13 months after diagnosis, mainly due to spinal tumor metastases, usually associated with delayed diagnosis of intracranial lesions.77 Provenzale JM, Gururangan S, Klintworth G. Trilateral retinoblastoma: clinical and radiologic progression. AJR Am J Roentgenol. 2004; 183(2):505-11. Some authors recommend routine assessment in patients with hereditary retinoblastoma with periodic MRI-SO until 4 years of age for early detection of asymptomatic tumors of the pineal or suprasellar region.88 Mouratova T. Trilateral retinoblastoma: a literature review, 1971-2004. Bull Soc Belge Ophtalmol. 2005; (297):25-35.
9 Amoaku WM, Willshaw HE, Parkes SE, Shah KJ, Mann JR. Trilateral retinoblastoma: a report of five patients. Cancer. 1996; 78(4):858-63.-1010 De Potter P, Shields CL, Shields JA. Clinical variations of trilateral retinoblastoma: a report of 13 cases. J Pediatr Ophthalmol Strabismus. 1994; 31(1):26-31.
-
Study conducted at Hospital A.C. Camargo, São Paulo, SP, Brazil
References
-
1Kivela T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009; 93(9):1129-31.
-
2Boubacar T, Fatou S, Fousseyni T, Mariam S, Fatoumata DT, Toumani S, et al. A 30-month prospective study on the treatment of retinoblastoma in the Gabriel Toure Teaching Hospital, Bamako, Mali. Br J Ophthalmol. 2010; 94(4):467-9.
-
3Knudson Jr AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971; 68(4):820-3.
-
4Kivelä T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999; 17(6):1829-37.
-
5Shields CL, Meadows AT, Shields JA, Carvalho C, Smith AF. Chemoreduction for retinoblastoma may prevent intracranial neuroblastic malignancy (trilateral retinoblastoma). Arch Ophthalmol. 2001; 119(9):1269-72.
-
6Bader JL, Meadows AT, Zimmerman LE, Rorke LB, Voute PA, Champion LA, et al. Bilateral retinoblastoma with ectopic intracranial retinoblastoma: Trilateral retinoblastoma. Cancer Genet Cytogenet. 1982; 5(3):203-13.
-
7Provenzale JM, Gururangan S, Klintworth G. Trilateral retinoblastoma: clinical and radiologic progression. AJR Am J Roentgenol. 2004; 183(2):505-11.
-
8Mouratova T. Trilateral retinoblastoma: a literature review, 1971-2004. Bull Soc Belge Ophtalmol. 2005; (297):25-35.
-
9Amoaku WM, Willshaw HE, Parkes SE, Shah KJ, Mann JR. Trilateral retinoblastoma: a report of five patients. Cancer. 1996; 78(4):858-63.
-
10De Potter P, Shields CL, Shields JA. Clinical variations of trilateral retinoblastoma: a report of 13 cases. J Pediatr Ophthalmol Strabismus. 1994; 31(1):26-31.
Publication Dates
-
Publication in this collection
Jul-Aug 2015
History
-
Received
13 Aug 2014 -
Accepted
24 Aug 2014