
SYNDROME IN QUESTION
Do you know this syndrome?* * Work done at the Dermatology Service, Santa Casa de Belo Horizonte
Paula de Rezende SalomãoI; PGustavo Carneiro NogueiraII; Maria Ester Massara CaféI
IMD, Assistant and Lecturer of the Pediatric Dermatology Ambulatory, Santa Casa de Belo Horizonte - Minas Gerais (MG)
IIDermatology specialist
Correspondence Correspondence to Paula de Rezende Salomão Rua Minas Nova, 215 / 301 - Cruzeiro 30310-090 - Belo Horizonte - MG
CASE REPORT
A five-year old male child with leukoderma was born and lives in Contagem, Minas Gerais state. He was born at full term in a normal pregnancy to nonconsanguineous parents, with appropriate weight and stature for this gestational age. The patient's family background was negative for congenital anomalies or genetic diseases.
The parents report that since birth, the patient showed spots on the dorsal aspect of the hands. Their size increased over time, and later appeared on the trunk.
The patient also had a finished face and hands, chest and lower limb deformities. The parents denied photosensitivity, hematomas and ulcerations on the exposed areas. The physical examination revealed the following alterations:
Skin: hypochromic and hyperchromic reticulated and atrophied spots in association with telangiectasias on the dorsal aspect of the hands and feet, knees, elbows, on the lateral side of the trunk and bilateral flanks;
Face: fine-tipped nose, discreet hypertelorism, micrognathism and craniofacial disproportion;
Trunk: thorax piriformis;
Limbs: deformity in flexing the metacarpophalangeal joints and knock-knees (geno valgo)
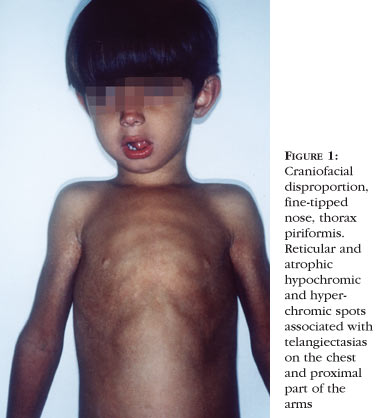
Oral lesions were not present nor were lesions of the skin and nails. Weight, stature and neuropsychomotor development were appropriate to age.
The skin biopsy of left lateral region of the trunk showed orthokeratosis and sinuous epidermis. The dermis was slightly atrophied, with discreet chronic inflammatory perivascular superficial infiltrate and vascular ectasias. Staining for elastic fibers, showed them to be reduced and fragmented.
The simple X-rays showed various types of alterations: craniofacial disproportion, widening of the sagittal, coronal and lambdoid sutures; minimum degree of micrognathism, hypertelorism and salience in the parietotemporal regions; thorax piriformis, thinning of the clavicles bilaterally with porosis of its distal extremity; slight alteration of the femoral tibial anatomic axis with valgus deformity; absorption of the most pronounced distal phalanges of the index fingers; multiple, lytic and focal lesions of the fine and sclerotic walls, localized in both distal femoral metaphyses.


WHAT SYNDROME IS IT?
Acrogeria (Gottron's Syndrome)
The clinical and histological findings of skin lesions associated with osteoarticular alterations were compatible with the diagnosis of acrogeria.
Acrogeria is one of the classic congenital premature ageing syndromes, among which are: pangeria (Werner's syndrome), progeria (Hutchinson-Gilford's syndrome) and acrogeria (Gottron's syndrome). In addition, various other diseases combine with premature ageing, like Cockayne's syndrome, Bloom's syndrome, telangiectasia (Louis-Bar's syndrome), Kindler's syndrome, trisomy 21 (Down's syndrome).1 These are rare genetic diseases associated with accelerated ageing of the skin and other tissues.2
Acrogeria was originally described by Gottron in 1941, when he observed premature cutaneous ageing localized on the hands and feet in two brothers, which were present ever since birth.3 Since then, various cases have been published, most in the European literature and predominantly affecting women. Most patients were of short stature, in spite of some being of normal height. Micrognathism is a frequent finding, and most of the cases described show atrophy of the skin at the tip of the nose, which gives a sculptural appearance. The registered mark of the condition is fine (atrophic) and wrinkled skin, with hyperchromia, telangiectasias and veins prominent on the upper trunk, and dorsal aspect of the hands and feet. The nails may be dystrophic or thick, but are usually normal.4-6
In the skin histopathology, there is atrophy of the dermis and subcutaneum. The collagen fibers are loose and dispersed, and the elastic fibers are always fragmented. The epidermis is spared.
In spite of some patients having clinical findings similar to those of progeria and metageria, they do not usually show generalized grave atherosclerosis. Therefore, they do are not usually have premature myocardic or coronary disease, which are the main causes of death. Life expectancy appears to be normal, with a risk of atherosclerosis in the general population.7
X-ray findings for most cases have received little emphasis in most of the published case reports. Due to this, a number of authors consider acrogeria mainly a cutaneous affection, but the bone alterations are well described as part of the syndrome.8 For patients who concomitantly show typical alterations of acrogeria and metageria, the single term of "Acrometageria" has been proposed, which can refer to the broadest spectrum of premature ageing syndromes. However, this concept is still not widely accepted in the medical literature.9 As these are very rare syndromes, all sharing an aspect of ageing skin similar to progeria, they are also at times called progeroid syndromes.10
The etiology of acrogeria is still not well elucidated. It has been considered autosomal dominant and autosomal recessive, though most reported cases have a positive family background. Mutations in the COL3A1 gene were identified in varied phenotypes, including acrogeria and vascular rupture in Ehlers-Danlos' syndrome.11,12 In the fibroblast culture a reduction of RNA messenger cells in collagen types I and II was found, as well as reduced life expectancy of the fibroblasts most prematurely showing morphological alterations typical of ageing. This is perfectly compatible with the patients' aged phenotype.13
There is currently no specific treatment available for either of these so-called progeroid syndromes. With this in mind, what is most important when making a differential diagnosis with them lies in the prognosis, which appears to be far better in acrogeria, as aforementioned. q
REFERENCES
1. Beauregard S, Giechrest BA. Syndromes of premature ageing. Dermatol Clin. 1987;5: 109-21.
2. Brown WT. Premature ageing syndromes. Curr Probl Dermatol. 1987;17: 152-65.
3. Gottron H. Familiare akrogerie. Arch Dermatol Syph. 1941;181: 571-83.
4. Calvert HT. Acrogeria (Gottron type). Br J Dermatol. 1957;69: 69.
5. Groot WP, Tafelkruyer J, Woerdeman MJ. Familial acrogeria (Gottron). Br J Dermatol.1970;103: 213.
6. Hjortshoj A, Heydernreich G. Acrogeria. A case report. Dermatologica. 1977;154: 355.
7. Lenane P, Krafchick B. Aging disorders. In: Schchner LA, Hansen RC, editors. Pediatric Dermatology. Mosby; 2003: 365-8.
8. Ho A, White SJ, Rasmussen JE. Skeletal abnormalities of acrogeria, a progeroid syndrome. Skeletal Radiol. 1987;16: 463-8.
9. Greally JM, Boone LY, Lenkey SG, Wenger SL, Steele MW. Acrometageria: a spectrum of "premature aging" syndromes. Am J Med Genet. 1992;44: 334-9.
10. Gilkes JJ, Sharvill DE, Wells RS. The premature ageing syndromes. Reports of eight cases and description of a new entity named metageria. Br J Dermatol. 1974;91: 243-62.
11. Pope FM, Narcisi P, Nicholls AC, Germaine D, Pals G, Richards AJ. COL3A! mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol. 1996;135: 163-81.
12. Jansen T, De Paepe A, Luytinck N, Plewig G. COL3A1 mutations leading to acrogeria (Gottron type). Br J Dermatol. 2000;142: 178-9.
13. Hashimoto C, Abe M, Onozawa N, Yokoyama Y, Ishikawa O. Acrogeria (Gottron type): a vascular disorder? Br J Dermatol. 2004;151: 497-501.
Publication Dates
-
Publication in this collection
13 June 2005 -
Date of issue
Apr 2005
