Abstracts
The polycystic ovary syndrome is an extremely common endocrine disorder in women of chilbearing age. It is characterized by menstrual disturbance, hyperandrogenism and/or hyperandrogenemia. The primary pathophysiological defect is unknown, but important characteristics include insulin resistance, androgen excess and impaired gonadotropin dynamics. The most frequent clinical characteristics of polycystic ovary syndrome are associated with the pilosebaceous unit, such as hirsutism, acne, seborrhea and alopecia. Thus, the dermatologist may be responsible for making an early diagnosis of the syndrome, thus preventing delay in establishing preventive and therapeutic measures. The current management recommended for skin manifestations of polycystic ovary syndrome includes combined oral contraceptives, antiandrogens and insulin-sensitizing agents, besides changes in life style. This is a review article on diagnosis, pathophysiology and treatment of polycystic ovary syndrome. The authors emphasize that a clear understanding of pathophysiology of this syndrome, especially by dermatologists, is crucial for its preventive treatment through the different phases in the life of women.
Hyperandrogenism; Hyperandrogenism; Skin; Insulin resistance; Hypothalamic-pituitary axis; Treatment
A síndrome do ovário policístico é distúrbio endócrino feminino, extremamente comum na idade reprodutiva. Caracteriza-se por anormalidades menstruais, hiperandrogenismo e/ou hiperandrogenemia. A principal alteração na fisiopatologia é desconhecida. Entretanto, parece que a resistência à insulina, o hiperandrogenismo e a alteração na dinâmica das gonadotropinas são os mais importantes mecanismos fisiopatológicos envolvidos. As características clínicas mais freqüentes da síndrome do ovário policístico estão relacionadas com a unidade pilossebácea, como hirsutismo, acne, seborréia e alopecia. Desse modo, o dermatologista pode ser responsável pelo diagnóstico precoce da síndrome, evitando o retardo na instituição de medidas terapêutico-preventivas. Atualmente, as drogas recomendadas para as manifestações cutâneas da síndrome do ovário policístico são os contraceptivos orais conjugados, antiandrógenos e sensibilizantes de insulina e, além disso, é geralmente recomendada a modificação no estilo de vida. Trata-se de artigo de revisão sobre diagnóstico, fisiopatologia e tratamento da síndrome do ovário policístico. Os autores enfatizam que o conhecimento da fisiopatologia dessa síndrome, principalmente pelos dermatologistas, é fundamental para seu tratamento preventivo, nas diferentes fases da vida da mulher.
Hiperandrogenismo; Hiperandrogenismo; Pele; Resistência à insulina; Sistema hipotálamo-hipofisário
REVIEW ARTICLE
Hyperandrogenism and skin: polycystic ovary syndrome and peripheral insulin resistance* * Work done at Universidade Federal de São Paulo - Escola Paulista de Medicina (Unifesp - EPM) - São Paulo (SP), Brazil.
Samira YarakI; Ediléia BagatinII; Karime Marques HassunIII; Meire Odete Américo Brasil ParadaIV; Sérgio Talarico FilhoV
Universidade Federal de São Paulo - Escola Paulista de Medicina (Unifesp-EPM) - São Paulo (SP), Brazil - Department of Dermatology
ICommissioned physician. Master's degree in Dermatology
IIPhysician. Ph.D. degree in Dermatology
IIIPhysician. Master's degree in Dermatology
IVVolunteer physician
VAdjunct Professor, Master's degree in Dermatology
Correspondence Correspondence Samira Yarak Departamento de Dermatologia Unifesp - Escola Paulista de Medicina Rua Botucatu, 740 04023-900 - São Paulo - SP Tel./fax: (11) 5571-2947 E-mail: sa.la@ terra.com.br
ABSTRACT
The polycystic ovary syndrome is an extremely common endocrine disorder in women of chilbearing age. It is characterized by menstrual disturbance, hyperandrogenism and/or hyperandrogenemia. The primary pathophysiological defect is unknown, but important characteristics include insulin resistance, androgen excess and impaired gonadotropin dynamics. The most frequent clinical characteristics of polycystic ovary syndrome are associated with the pilosebaceous unit, such as hirsutism, acne, seborrhea and alopecia. Thus, the dermatologist may be responsible for making an early diagnosis of the syndrome, thus preventing delay in establishing preventive and therapeutic measures. The current management recommended for skin manifestations of polycystic ovary syndrome includes combined oral contraceptives, antiandrogens and insulin-sensitizing agents, besides changes in life style. This is a review article on diagnosis, pathophysiology and treatment of polycystic ovary syndrome. The authors emphasize that a clear understanding of pathophysiology of this syndrome, especially by dermatologists, is crucial for its preventive treatment through the different phases in the life of women.
Keywords: Hyperandrogenism / diagnosis; Hyperandrogenism / therapy; Skin; Insulin resistance; Hypothalamic-pituitary axis; Treatment
INTRODUCTION
Hyperandrogenism is a term used to describe the clinical signs related to the biological action of androgens.1-3 Hyperandrogenemia or biochemical hyperandrogenism means increased blood androgen levels.2 The maximum clinical expression of hyperandrogenism is virilization.2 Idiopathic hyperandrogenism is characterized by clinical expression and no biochemical alterations, whereas occult hyperandrogenism is characterized by no clinical expression and presence of biochemical changes.2
Hyperandrogenism leads to clinical pictures of variable severity in females, including early puberty, hirsutism, acne, seborrhea, alopecia, menstrual disturbance and ovulatory dysfunction with infertility during reproductive life, metabolic syndrome, psychological disorders and virilization.1,2,4The intensity and extension of these clinical manifestations depend on several factors,1 1 Etiology, sex, age, association with other hormone disorders and individual susceptibility factors. 2 and there is no strict correlation between intensity of clinical picture and biochemical alterations.2-4
Several etiologies may cause hyperandrogenism in women, ranging from hormone disorders2,5 in the ovaries and adrenal glands (polycystic ovary syndrome - PCOS and non-classical congenital adrenal hyperplasia - NCCAH) to ovarian or adrenal cancer.2
The main form of hyperandrogenism in females is functional ovarian or PCOS,6-9 which accounts for two-thirds of hyperandrogenic women,2,6 and approximately 50% of PCOS cases present functional adrenal hyperandrogenism.2,4,9
PCOS has a very heterogeneous clinical picture4,6-9 and is the most common endocrinopathy in women of childbearing age,10 with a prevalence of 6% to 10%.6,11,12 It is characterized by clinical and/or biochemical hyperandrogenism and menstrual irregularities,10 and it is probably the most frequent cause of hirsutism and infertility.6 The etiopathogenesis is still unknown,7,11 although several associations with biochemical abnormalities have been described.6,7,13
Stein, Leventhal14 were the first investigators to identify the association of enlarged and polycystic ovaries and amenorrhea, hirsutism and obesity. PCOS was associated with adrenal hyperplasia8,15 and, later, with hyperinsulinemia,16 thus demonstrating its metabolic and reproductive effects. Other investigators17-19 observed later that the defect was in insulin receptors and obese and non-obese women presented hyperinsulinemia.20 From then on, the association between hyperandrogenism, hyperinsulinemia21,22 and alterations in serum lipid levels23-25 was given special attention for being able to change the prognosis of PCOS.23
Many factors contribute to making diagnosis of PCOS difficult. The National Institute of Child Health & Human Development (NIHD/NICHD)26 considered the following diagnostic criteria for PCOS: presence of chronic anovulation, hyperandrogenism and/or hyperandrogemia and ruling out other endocrine diseases with similar phenotype. The European Society of Human Reproduction and Embryology and the American Society for Reproductive Medicine9 (ESHRE/ASRM) recommended, apart from ruling out other conditions, the presence of two out of three criteria, as follows: 1) menstrual dysfunction with oligo-ovulation or anovulation, 2) hyperandrogenism or hyperandrogenemia and 3) presence or not of polycystic ovaries.
In principle, the early diagnosis of PCOS is important for the possibility of preventing the development of some diseases associated with the syndrome. Thus, dermatologists may be responsible for making such diagnosis due to the skin manifestations of hyperandrogenism. The objective of this review is to provide an update on pathophysiology, diagnosis and treatment of PCOS, considering that, in the past 10 years, the pathophysiological and therapeutic aspects have been correlated, particularly regarding peripheral insulin resistance.
HYPOTHALAMUS-PITUITARY-OVARY-ADRENAL AXIS - ANDROGEN BIOSYNTHESIS, FACTORS RELATED TO ITS PRODUCTION AND THE ROLE OF INSULIN
1. Hypothalamus-pituitary-ovary-adrenal axis
The hypothalamus and the pituitary gland are the structures that regulate the endocrine system.5 The sensorial and endocrine information is processed and integrated in the brain, by means of connections between the pituitary gland and hypothalamic or portal system neurons.3,5In the pituitary anterior lobe, the portal system produces peptides that bind to specific cell membrane receptors, thus initiating hormone release (HR) or inhibition (HI)2 2 Positive and negative feedback mechanisms. (Chart 1).3,5,27,28 The hypothalamus stimulates the production of gonadotropins in the pituitary through pulsatile gonadotropin-release hormone (GnRH), increasing the transcription of gonadotropin genes (luteinizing hormone - LH and follicle-stimulating hormone - FSH) (Figure 1A).27,28 Hence, the frequency of pulsatile GnRH stimulus partly determines the relative proportion LH and FSH synthesis.27 The increased frequency of GnRH pulse favors the transcription of LH b -subunit over FSH; conversely, decreased GnRH pulse frequency favors the transcription of FSH b-subunit, reducing the transcription rate of LH over FSH.27,28
2. Androgen biosynthesis and factors related to its production
The androgens derive from cholesterol and, in females, are synthetized by the ovaries, adrenal glands and in extraglandular sites of steroid conversion (liver, muscles, skin and adipose tissue).29,30 Androgen aromatization occurs in muscle and adipose tissues, that is, testosterone (T) and androstenedione (A) are converted into estrogens - estrone and estradiol, whereas, in the pilosebaceous unit and skin, T is converted into dihydrotestosterone (DHT) by the enzyme 5-alpha-reductase 1 or 2 (Figure 1B).29
The pilosebaceous unit and skin represent target-structures for androgen, which explains the pathophysiology of hyperandrogenism cutaneous manifestations (hirsutism, acne, seborrhea and alopecia).2
3-alpha-androstenediol glucuronide (3a diol G) derives from the conversion of DHT and A, by means of 5a-redutase. It is considered a marker of androgen biological action in the pilosebaceous unit, and the skin its main production site.2,4,29,30
Androgen biosynthesis (Figure 2) is mediated by cytochrome P-450c-17, an enzyme with 17a-hidroxylase, 17, 29-lyase and 17b-hydroxysteroid dehydrogenase (17b HSD or 17b R) activities. The androgens (A and T) are aromatized to estrone by the enzyme aromatase (cytochrome p-450 aromatase).27,29,31
In the ovaries, the androgens are precursors of estrogens and their production is controlled by LH/FSH (Figures 1A and 2, and Chart 2).29,30,32 Normal ovarian function is determined by a combined action of LH in the theca cells, corpus luteum and stroma, and of FSH in granulosa cells.28
FSH stimulates the synthesis of estrogens, inhibin, activin and follistatin in granulose cells.33 SHBG, IGF, inhibin, activin and follistatin release by granulose cells modulate the amount of androgens made in response to LH.34-36 Insulin and the insulin-like growth factor (IGF) enhance the action of FSH in granulosa cells (Figure 1A).13,31,32,34
Eighty percent of circulating T is bound to a protein produced by the liver - b-globulin (SHBG), 19% is bound to albumin, and only 1% is free and responsible for the peripheral effect of androgens.3,29,30
Increased levels of b-globulin are related to higher levels of estrogens and thyroid hormones, while androgens, obesity, glucocorticoids, growth hormone (GH) and insulin inhibit its synthesis.3,29
Androgen binding to androgen receptors (AR) is related to DNA, production of mRNA and of enzyme proteins required for its action (Figure 1B).4,31
3. The role of insulin
Insulin is a polypeptide secreted by b-cells of the pancreas, and play an important role in glucose homeostasis.4,19,31 The classic target tissues include liver, muscles and adipose tissue. The terms insulin sensitivity and insulin resistance (IR) refer to the action of insulin in glucose homeostasis.4,19,31
PATHOPHYSIOLOGY OF THE PCOS
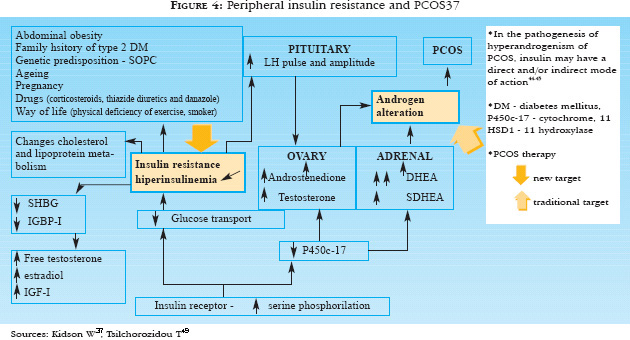
The heterogeneity of PCOS reflects the participation of multiple pathophysiological mechanisms (Figure 3); however, how much each mechanism contributes to developing PCOS is still unknown.
Biochemical abnormalities13 were described and it seems that the primary defect is IR in muscles and adipose tissue, associated with compensatory hyperinsulinemia although the ovaries remain sensitive to insulin.19,37-39 Furthermore, IR may be related to an intrinsic dysfunction of pancreatic b-cells.38
The genetic etiology may be observed in mothers and sisters of PCOS patients, mainly because of higher frequency of the syndrome40 and IR,41 Nevertheless, the mode of inheritance remains uncertain and unknown, as well as the influence of several environmental factors, such as diet and life style.
Another important characteristic in PCOS is changes in gonadotropin metabolism.4,9In vivo and in vitro studies (thecal cell culture) in women with PCOS suggested that thecal cells are more efficient to convert androgen precursors into T than normal cells. The capacity of these cells to increase production of androgen is intrinsic and it might be genetically determined.29,30 Other in vivo studies42,43 observed abnormalities in the granulose cells of PCOS patients.
The theories proposed to explain the pathophysiology of PCOS could be divided into four categories:
a) single defect in action and secretion of insulin, causing hyperinsulinemia and IR;
b) primary neuroendocrine defect, leading to increased pulse frequency and amplitude of LH;
c) defect in androgen synthesis, resulting in increased production of ovarian androgens; and
d) alteration in cortisol metabolism, resulting in increased production of adrenal androgens.
1. Hyperinsulinemia and IR
Hyperinsulinemia is believed to be a biochemical, central and, probably hereditary alteration of PCOS (Figure 4). In 1980, the association between hyperandrogenism, insulin resistance and acanthosis nigricans was identified (Hairan syndrome).16 Since then IR has been the best biochemical correlation for acanthosis nigricans.4
Insulin may act through insulin receptors, which are distributed in the ovaries,44,45 through IGF-1 receptors or even by means of hybrid receptors that contain a combination of a and b ubunits of insulin and IGF-1 receptors.46 Apparently there is a hereditary genetic predisposition in post-receptor mechanisms of insulin, for many obese and insulin-resistant women do not develop PCOS.19,46
The tyrosine-kinase insulin receptors include IGF-1, epidermal growth factor, fibroblast growth factor, platelet-derived growth factor, colony-stimulating factor-1 and several cytokine receptors.18,19 The factor that induces phosphorylation of serin in insulin receptor and in cytochrome P450-c17a seems to be a protein-kinase, which causes IR and hyperandrogenism, respectively. The factor responsible for serin phosphorylation is genetically determined. Therefore, the genetic defect in serin phosphorylation could explain the association of PCOS and IR.19,24 Hyperinsulinemia increases the production of androgens in the ovaries and of IGFs in the liver.37,39 The direct effect of insulin and IGF-1 is increased 17-hydroxilase activity in the ovaries, causing an excessive production of androgens, particularly A and T and its precursor, 17-hydroxyprogesterone (17-OHP).46 IGF-1 inhibits the enzyme aromatase and hence prevents the conversion of T into estrogens.47 Indirectly, insulin seems to potentialize the action of LH in the ovaries.48 Another effect of hyperinsulinemia, and similar to obesity, is to decrease the hepatic production of SHBG, the protein that carries the sexual hormone sexual, and of IGFBP-1, the protein that carries IGF-1 or IGF binding protein-1, thus contributing to a broader action of free testosterone (FT) and IGF-1, respectively in target-cells.45,49
Glucose, leptin and lipid intolerance
PCOS is a risk factor more important for glucose intolerance than race ethinicity.24 The association between PCOS and gestational diabetes (type 1 and 2I) in families is controversial.50,51
Leptin3 3 The obese individuals present leptin resistance. 52, 53 seems to have a direct effect in ovarian steroidogenesis due to the presence of leptin receptors in thecal and granulose cells; moreover, granulose cells are able to synthesize leptin (Figure 1A).52-54 Peripheral insulin resistance probably accounts for reduction in leptin55 and resistin56 concentration in the adipocytes of women with PCOS.
In PCOS, total cholesterol is increased because of raised low-density lipoprotein (LDL); however, high-density lipoprotein (HDL) is decreased. Blood levels of triglycerids are elevated, as well as plasminogen activator inhibitor (PAI). The increase in PAI levels and alteration in lipid levels seem to be responsible for the increased incidence of hypertension, coronary artery disease and thrombosis in PCOS.57
2. Defect in the neuroendocrine system
By and large, the inappropriate secretion of gonadotropins is associated to the classical PCOS. The increased secretion of gonadotropins is related to increased activity of GnRH pulse generator and to pituitary response to GnRH. LH and FSH synthesis and secretion depend on GnRH stimulus, which is characterized by fast and slow frequencies that, respectively, favor their secretion.58 Deregulation of the mechanism of GnRH secretion is still not clear. Through observations in the peripubertal period (adrenarche) in females, it was suggested that changes in neuronal system information caused by insulin, IGFs (IGF1 and IGF2) and sexual steroids could induce deregulation of GnRH pulse generator.42,59 The weak peripheral aromatization of the androgen from A and estrone concentration may increase pituitary sensibility to GnRH by direct action in the synthesis of gonadotropins, as well as sensibility of GnRH receptors,60 cthus contributing to pathogenesis of PCOS and justifying the increased LH levels and the excessive response of LH to GnRH. Marshall et al.58 observed some abnormalities in the function of hypothalamic neurotransmitters.4 4 Dopamine, opioids and the alpha-adrenergic system. However, it is still not clear if these neurotransmitters play any role in the pathophysiology of PCOS. In the polycystic ovary syndrome, during the follicular phase, there is a disproportion of gonadotropins, that is, LH pulsatile secretion is increased and that of FSH is reduced.60The increase in the LH/FSH ratio5 5 The prevalence of increase in this ratio ranges from 30% to 90%. Such variability is due to the number and interval of samples collected and to specificity of gonadotropins. The gene responsible for specificity is in LH b-subunit, and mutations in this subunit might be associat-ed with gonadal dysfunction and infertility. 61- 63 from 2:1 to 3:1 indicates the abnormal secretion of gonadotropins.61-63
3. Defect in ovarian steroid synthesis
PCOS patients present an increase in GnRH28 and LH,64 pulse frequency and, consequently, in ovarian androgen synthesis.28,29,49,64 It is still unknown if increased GnRH pulse frequency is due to intrinsic abnormalities in GnRH pulse generator or if it caused by relatively low levels of progesterone, resulting in anovulatory cycles (Figure 3).28,29
Some studies carried out thecal cell cultures suggest that these cells, in PCOS, are more efficient to convert androgen precursors into T than the normal cells,29 and there is also deregulation in ovarian steroidogenesis due to changes in the enzymes 17-hydroxylase and 17, 29-lyase, that is, in cytochrome P450c of thecal cells.28,30,49 Moreover, the synergic effect of hyperinsulinemia and the increase in IGF must be considered.48,49,65
In women suffering from PCOS, follistatin levels are elevated and activin levels are lower than in non-PCOS women.66
The peripheral metabolism of steroids is altered in PCOS, primarily in adipose and muscular tissues and in the pilosebaceous unit. Hence, hirsutism, acne, seborrhea and alopecia are common and reflect hyperandrogenism, which may be progressive or not.
The adipose tissue is able to form T and estrone from inactive precursors,67 contributing to increased steroid levels. In the pilosebaceous unit, there is an increase in activity of the enzyme 5a-redutase, reductase, converting T into DHT.68,69 The activity of 5areductase is mediated by IGF-1 and may be intensified by hyperinsulinemia, thus aggravating hirsutism.68,69 Insulin seems to have a direct and stimulating effect in the pilosebaceous unit (hirsutism, acne, seborrhea and alopecia)4 and in the epidermis (acanthosis nigricans).16
4. Peripheral increase in cortisol metabolism
Increased androgen production by adrenal glands is observed in 25% of PCOS patients,70 probably as a result of genetic influence or secondary to abnormal secretion of ovarian androgens.31,65
GENETICS OF PCOS
PCOS is believed to be hereditary; however, the mode of inheritance is unknown. The several genes4,19,29,30,40,42,43 proposed and investigated as the main and possibly PCOS-related genes include those that regulate the hypothalamus-pituitary-ovary axis29,30,42,43 and those associated with peripheral insulin resistance and its sequelae.4,19,40,41 New genes have been identified and are related to insulin and gonadotropin action and secretion, and androgen biosynthesis, secretion, transport and metabolism, contributing to the heterogeneous phenotype of this syndrome.64,66,71
CLINICAL PICTURE AND DIAGNOSIS OF PCOS
The clinical picture (Chart 3) and the heterogeneous hormone profile, the multifactorial theory of its pathogenesis and lack of consensus as to definition of PCOS represent the factors that contribute to making its diagnosis difficult.
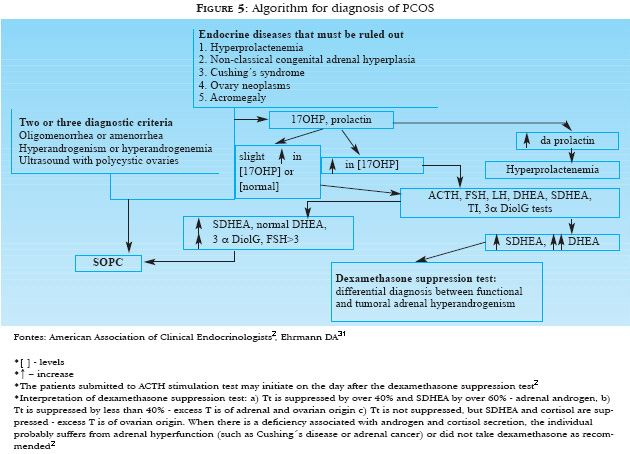
According to the last consensus,9 a SOPC PCOS could be diagnosed after ruling out other diseases that cause irregular menstrual cycles and androgen excess (Figure 5, Chart 4), by means of identifying at least two of the following criteria: oligo-ovulation or anovulation, which is usually manifested with oligomenorrhea or amenorrhea; increased androgen levels in blood (hyperandrogenemia) or clinical manifestations of androgen excess (hyperandrogenism); and ovary cysts defined by ultrasonography (10 or more cysts measuring 2-8mm).9
PCOS is a chronic condition that has clinical manifestations clinical at any age and not only in women of childbearing age (Chart 3).71 The classical form is characterized by high androgen, estrogen, insulin and LH levels. The metabolic syndrome (Chart 5)9 in patients with PCOS, especially in premenopausal women, is characterized by obesity, IR and dyslipidemia. The increase in triglycerid and total cholesterol levels, with elevated LDL and reduced HDL,25,72 characterizes dyslipidemia. Hirsutism6 6 Hirsutism is defined as the excessive growth of terminal hair in women, in characteristic anatomical areas, which are androgen-sensitive or with a male distribution pattern. In hirsutism there is an excessive or inappropriate development and growth of the pilosebaceous unit. The androgens cause transformation of vellus hair (which is fine, soft and non-pigmented) into terminal hair in androgen-sensitive areas. 2,72 is the most frequent clinical feature of hyperandrogenism in skin and it may cause psychiatric disorders in some patients.
It is important to make an early diagnosis of PCOS because of its association with high reproductive morbidity and increased risk of developing hormone-dependent cancer, thus justifying a preventive treatment.23 Diabetes mellitus type 2, dyslipidemia, endometrial cancer, hypertension, cardiovascular disease and ovary cancer are long-term risk factors.23,25,72
The complementary exams required for PCOS investigation are basically ultrasound and hormone levels.2,9,7
1. Ultrasonography
It should be performed between day 25 of the current menstrual cycle and day 3 of the next period. Transvaginal, rather than abdominal, ultrasound is recommended for providing better visualization2,61
2. Hormones
General orientations
a) Collect blood in the morning (8h), in the first week of the regular menstrual cycle.7 7 Irregular menstrual cycles: random blood collection.
Plasma hormone levels
a) 17-OHP - it is important to perform this test in the morning because of corticotropin peak. In PCOS, 17-OHP levels may be normal or slightly increased.31 The plasma prolactin and 17-OHP levels are enough to rule out hyperprolactenemia and NCCAH due to 21-hydroxylase deficiency.
b) Prolactin - normal levels.31
c) FSH and LH - LH/FSH ratio is > 3:1.61
d) TT, FT and SHBG - the method recommended9 to calculate FT is by measuring TT and SHBG levels. FT levels are increased and SHBG levels are reduced. TT levels may be elevated or normal.2,9
e) Dehydroepiandrosterone DHEA and SDHEA - both in PCOS and NCCAH, the increase in SDHEA levels is lower than 7mg/dL. In adrenal tumors, SDHEA is higher than 7mg/dL. DHEA is increased in NCCAH (Chart 2).2
f) 3-alfa diol G - it is a DHT metabolite produced in androgen-responsive tissues (for instance, hair follicle). It is raised in PCOS, idiopathic hirsutism and it is decreased in 5a-reductase deffciency.2
g) Adrenocorticotropic hormone (ACTH) stimulation test. Cortrosyn® - 250ug IV- the test is performed to detect deficiency of several steroidogenesis enzymes in adrenal glands, mainly 21-hydroxylase deficiency.2 The test is indicated only for screening morning levels of 17-OHP with above normal concentrations or if there is any suspicion of enzyme deficiency. Plasma cortisol, 17-OHP, SDHEA and DHEA should be measured in the morning, before and 60 minutes after Cortrosyn® injection.2
h) GnRH stimulation test - it is conducted to confirm the ovarian origin of androgens. In PCOS investigation it is useful to identify the presence of 17- hydroxylase deficiency.2
i) Dexamethasone suppression test (Figure 5) - it is performed to differentiate functional adrenal hyperandrogenism (ACTH-dependent) from tumoral adrenal hyperandrogenism (ACTH-independent). The test is indicated when TT is higher than 200ng/mL, and/or SDHEA is higher than 7mg/dL. Plasma cortisol, SDHEA and TT should be measured in the morning before and five days after the administration of dexamethasone (0.5mg, four times/day, at 6, 12, 18 and 24 hours).2,8 8 The decrease in androgen levels may occur by inhibiting its production in the ovaries, by increased SHBG levels or blocking receptors in target tissues.
j) IR - several criteria have been suggested to assess IR (Chart 5):72-75
· fasting glucose,
· body mass index (BMI), IR with BMI > 25kg m»,
· glycemic index,
· glucose tolerance test - 2 hours after 75mg of glucose,
· fasting insulin - IR with insulinemia > 17.3 UI/l in women,
· fasting glucose/insulin ratio,
· HOMA-r,
· HOMA-b analysis,
k) as to lipid metabolism, it is recommended to measure total cholesterol total and its fractions (HDL and LDL) and triglycerids (Chart 5).2
TREATMENT
The choice of treatment in PCOS depends on the clinical and laboratory pictures. In Dermatology, the objective of treating PCOS is to reduce androgen levels,8 8 The decrease in androgen levels may occur by inhibiting its production in the ovaries, by increased SHBG levels or blocking receptors in target tissues. in order to attenuate their effects on skin and pilosebaceous units. Together with drug therapy, it is necessary to change life style, diet and to exercise.76
The therapeutical options for skin manifestations related to androgen excess include, as follows:
1. combined oral contraceptives: cyproterone acetate (2mg) + ethinyl estradiol (35 mg) for 21 days or drospirenone9 9 Drospirenone is a spironolactone analogue. (3mg) + ethinyl estradiol (30mfor 28 days. The antiandrogen effect is due to decreased LH and, consequently, reduced ovarian production of androgen by the estrogenic component.2 Estrogens also increase the synthesis of SHBG, thus reducing FT.2 The use of oral contraceptives requires special monitoring, that is, gynecologic and breast examination.2 The effectiveness of contraceptives is greater in acne (50%) and lower in hirsutism, which requires administration for longer periods.2,31,77 he adverse effects of contraceptives in IR, glucose tolerance, vascular reactivity and coagulability should be taken into account, particularly after availability of drugs that reduce insulin levels.78
2. antiandrogens: a) cyproterone acetate (25mg to 50mg, in the first 10 days of the cycle) - it is a potent progesterone with moderate antiandrogen action. It inhibits T and DHT binding to androgen receptor, reduces the concentration of 5a reductase in the skin and decreases the secretion of ovarian androgens by inhibiting gonadotrofin release.2 Hirsutism improves after a 3-to-6-month treatment.77 The side effects include irregular uterine bleeding, nausea, vomiting, headache, fatigue, weight gain and reduced libido.2,79
b) spironolactone - it has a moderate antiandrogen effect when administered as a 100-200mg/day dose. Shaw80 published the following guidelines for spirolanoctone: initial dose of 25mg, twice a day; if well tolerated, increase to 50mg, twice a day; in case of no benefits after three months, higher doses should be considered. The duration of treatment could be extended and associated with combined oral contraceptives.
c) flutamide - potent nonsteroidal antiandrogen that is effective in treating hirsutism, but severe hepatocellular dysfunction has limited its use.31
d) finasteride - It is a 5a-reductase inhibitor that is given for hirsutism, 5mg/day, and patients improve after three-months81
e) eflornithine - - inhibitor of the enzyme decarboxylase ornithine in human skin, it is available in creams for topical use. The primary action of the drug is to inhibit hair growth.82
3. insulin-sensitizing agents: metformin, thiazolidinedione.31,78 These agents enhance tissue sensitization to the action of insulin.
a) metformina10 10 It is a biguanide molecule. - Velásquez83 published the first study on metformin in PCOS. Metformin reduces hepatic gluconeogenesis and increases muscle sensitization to insulin, decreasing serum insulin levels and thus reducing androgen production by thecal cells.84 However, it is not a hypoglycemic agent because it does not increase insulin secretion.2,85 Moreover, metformin reduces total cholesterol, LDH and triglycerid levels, and increases HDL levels.86,87 Decreased androgen concentration caused by metformin is an effect still not clear.84 Apparently it has direct influence in ovarian steroidogensis, reducing androgen production.84 he recommended dose is 500mg, 3x/day or 850mg, 3x/day. It is effective in treating anovulation in women with PCOS and, in long-term treatments it may improve hirsutism.87 The most important side effects are gastrointestinal symptoms (diarrhea, nausea, vomiting, flatulence and anorexia), which occur in 30% of patients, decreased B12 vitamin levels in 6% to 9%, and metallic taste in 3%. The contraindications are renal disease, metabolic acidosis, congestive heart failure, and hypersensitiveness to metformin. Avoid use of contrast medium containing iodine.2,83,84
b) thiazolidinediones (troglitazone, pioglitazone, rosiglitazone) - these are true insulin-sensitizing agents that are able to improve insulin action in the liver, skeletal muscles and adipose tissue.85,86 These drugs have the capacity to activate certain genes involved in fat synthesis in carbohydrate metabolism, thus improving IR, hyperandrogenemia and glucose tolerance,85-87 and they lead to less weight loss than other insulin-sensitizing agents.88 Roziglitazone and pioglitazone are currently approved for use. Treatment with roziglitazone was associated with improved function of pancreatic b-cells and decrease in PAI.85,86
c) D-chiro-inositol - it was recently studied in PCOS patients. It is a pure substance called pinitol,11 11 This drug is chemically defined as inositol. derived from pine trees.89 The therapy with D-chiro-inositol, in obese women with PCOS improves insulin metabolism by acting in its receptors, decreasing serum insulin and androgen (FT and increased SHBG) levels.86,89 he recommended dose is 1200mg/day.85
The response to therapy with insulin-sensitizing agents is directly proportional to BMI, fasting insulin, total cholesterol and LDL-cholesterol and blood pressure, and inversely proportional to A and HDL levels.
CONCLUSION
PCOS is a complex disorder of unknown etiology, and it involves several specialists for presenting reproductive, endocrinologic, dermatological, gynecological, cardiac and psychological implications.
Hyperinsulinemia seems to be one of the main factors responsible for steroidogenesis deregulation.
The variable and heterogeneous clinical picture makes diagnosis of PCOS difficult and tends to delay management that could avoid late complications.
Its treatment is preventive and aims to maintain the endometrial healthy, to antagonize the actions of androgens in target-tissues, to reduce insulin resistance (IR) and to correct anovulation. In addition to combined contraceptives and antiandrogens, the insulin-sensitizing agents are effective in preventing diseases associated with hyperinsulinemia. It is difficult to explain the therapeutic success of metformin in reducing insulin and androgen levels, as observed in some studies. It may be related to genetic variations, body weight, life style, duration of treatment and dosage of the drug.
Today, in order to avoid late complications, the specialists share investigations, trying to understand the etiology and pathophysiology of PCOS, which are essential for its treatment. The main focus of these studies has been several genetic and environmental determinants of the syndrome, for reflecting its heterogeneous phenotype.

REFERENCES
Received on May 31, 2005.
Approved by the Consultive Council and accepted for publication on July 04, 2005.
- 1. Spinedi E, Mariani V, Bulfon M, Colombani-Vidal M, Scaglia H. Analysis of the hypothalamic-pituitary-ovary axis in the neonatally-androgenized female rat. J Endocrinol Invest. 1990;13:481-8.
- 2. American Association of Clinical Endocrinologists. Medical guidelines for clinical practice for the diagnosis and treatment of hyperandrogenic disorders. Endocr Practice. 2001;7:121-34.
- 3. Perez Gutierrez JF. The physiology of the hypothalamo-hypophyseal axis. An R Acad Nac Med. 1994;111:333-61.
- 4. Rosenfield RL. Polycystic ovary syndrome and insulin-resistant hyperinsulinemia. J Am Acad Dermatol. 2001;45:S95-104.
- 5. Herman JP, Prewitt CM, Cullinan WE. Neuronal circuit regulation of the hypothalamo-pituitary-adrenocortical stress axis. Crit Rev Neurobiol. 1996;10:371-94.
- 6. Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metabol. 2004;89:2745-9.
- 7. Lobo RA, Carmina E. The importance of diagnosing the polycystic ovary syndrome. Ann Int Med. 2000;132:989-93.
- 8. Gallagher TF, Kappos A, Hallman L, Lipsett MB, Pearson OH, West CD. Adrenocortical hyperfunction in idiopathic hirsutism and the Stein-Leventhal syndrome. J Clin Invest. 1958;57:794.
- 9. The Rotterdam ESHRE/ASRM-sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long term health risks related to polycystic ovary syndrome (PCOS) Human Reprod. 2004;19:41-47.
- 10. Hull MG. Epidemiology of infertility and polycystic ovarian disease: endocrinological and demographic studies. Gynecol Endocrinol. 1987;1:235-45.
- 11. Polson DW, Wadsworth J, Adams J, Franks S. Polycystic ovaries - a common finding in normal women. Lancet. 1988;1:870-2.
- 12. Glueck CJ, Wang P, Goldenberg N, Sieve-Smith L. Pregnancy outcomes among women with polycystic ovarian syndrome treated with metformin. Human Reprod. 2002;17:2858-64.
- 13. Michelmore KF, Balen AH, Dunger DB, Vessey MP. Polycystic ovaries and associated clinical and biochemical features in young women. Clin Endocrinol. 1999;51:779-86.
- 14. Stein IL, Leventhal ML. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol. 1935;29:181-91.
- 15. Pinheiro AS, Clapauch R. Importância da dosagem da 17OH-Progesterona na síndrome dos ovários policísticos. Arq Bras Endocrinol Metab. 2001;45:361-8.
- 16. Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab. 1980;50:113-6.
- 17. Dunaif A, Hoffman AR, Scully RE, Flier JS, Longcope C, Levy LJ, et al. Clinical, biochemical, and ovarian morphologic features in women with acanthosis nigricans andmasculinization. Obstet Gynecol. 1985;66:545-52.
- 18. Dunaif A, Xia J, Book CB, Schenker E, Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest. 1995;96:801-10.
- 19. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18:774-800.
- 20. Chang RJ, Nakamura RM, Judd HL, Kaplan SA. Insulin resistance in nonobese patients with polycystic ovarian disease J Clin Endocrinol Metab. 1983;57: 356-9.
- 21. Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c-17a activity and serum free testosterone after reduction in insulin secretion in women with polycystic ovary syndrome. N Engl J Med.1996; 335:617-6.
- 22. Nestler JE, Jakubowicz DJ. Lean women with polycystic ovary syndrome respond to insulin reduction with decreases in ovarian P450c-17a activity and serum androgens. J Clin Endocrinol Metab. 1997;82:4075-9.
- 23. Sir-Petermann T, Maliqueo M, Pérez-Bravo F, Angel B, Carvajal F, Del Solar MP, et al. PCOS: the importance of establishing diagnosis. Rev Med Chil. 2001;129:805-12.
- 24. Legro RS, Kunselman AR, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84:165-9.
- 25. Birdsall MA, Farquhar CM, White HD. Association between polycystic ovaries and extent of coronary artery disease in women having cardiac catheterization. Ann Intern Med. 1997;126:32-5.
- 26. Zawadzki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens JR, Haseltine F, Merriam GR, editors. Polycystic ovary syndrome. Boston, MA: Blackwell; 1992. p. 377-84.
- 27. Haisenleder DJ, Dalkin AC, Ortolano GA, Marshall JC, Shupnik MA. Pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo Endocrinology. 1991;128:509-17.
- 28. Turgeon JL, Kimura Y, Waring DW, Mellon PL. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol Endocrinol. 1996;10:439-50.
- 29. Nelson VL, Qin Kn KN, Rosenfield RL, Wood JR, Penning TM, Legro RS, et al. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86:5925-33.
- 30. Nelson VL, Legro RS, Strauss JF 3rd, McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13:946-57.
- 31. Ehrmann DA. Polycystic ovary syndrome. N Engl J Med. 2005;352:1223-36.
- 32. Adams J, Franks S, Polson DW, Mason HD, Abdulwahid N, Tucker M, et al. Multifollicular ovaries: clinical and endocrine features and response to pulsatile gonadotropin releasing hormone. Lancet. 1985;2:1375-9.
- 33. Vale W, Wiater E, Gray P, Harrison C, Bilezikjian L, Choe S. Activins and inhibins and their signaling. Ann NY Acad Sci. 2004;1038:142-7.
- 34. Welt C, SidisYl, Keutmann H, Schneyer A. Activins, Inhibins, and Follistatins: Endocrinology to Signaling. A paradigm for the new millennium. Exp Biol Med. 2002;227:724-52.
- 35. Smitz J, Cortvrindt R, Hu Y,Vanderstichele H. Effects of recombinant activin A on in vitro culture of mouse preantral follicles. Mol Reprod Dev. 1998;50:294-304.
- 36. Li R, Phillips DM, Mather JP. Activin promotes ovarian follicle development in vitro Endocrinology. 1995;136:849-56.
- 37. Kidson W. Polycystic ovary syndrome: a new direction in treatment. Med J Aust. 1998;169:537-40.
- 38. Goodarzi MO, Erickson S, Port SC, Jennrich RI, Korenman SG. b-cell function: a key pathological determinant in polycystic ovary syndrome. J Clin Endocrinol. Metab. 2005;90: 310-15.
- 39. Rosenfield RL. Ovarian and adrenal function in polycystic ovary syndrome. Endocrinol Metab Clin North Am. 1999;28:265-93.
- 40. Kahsar-Miller MD, Nixon C, Boots LR, Go RC, Azziz R. Prevalence of polycystic ovary syndrome (PCOS) in first-degree relatives of patients with PCOS. Fertil Steril. 2001;75:53-8.
- 41. Yildiz BO, Yarali H, Oguz H, Bayraktar M. Glucose intolerance, insulin resistance, and hyperandrogenemia in first degree relatives of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:2031-6.
- 42. Coffler MS, Patel K, Dahan MH, Malcom PJ, Kawashima T, Deutsch R, et al. Evidence for abnormal granulosa cell responsiveness to follicle-stimulating hormone in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:1742-7.
- 43. Apter D, Butzow T, Laughlin GA, Yen SS. Accelerated 24-hour luteinizing hormone pulsatile activity in adolescent girls with ovarian hyperandrogenism: relevance to the developmental phase of polycystic ovarian syndrome. J Clin Endocrinol Metab. 1994;79:119-25.
- 44. Poretsky L, Smith D, Seibel M, Pazianos A, Moses AC, Flier JS. Specific insulin binding sites in human ovary. J Clin Endocrinol Metab. 1984;59:809-11.
- 45. Poretsky L, Cataldo NA, Rosenwaks Z, Giudice LC. The insulin-related ovarian regulatory system in health and disease. Endocr Rev. 1999;20:535-82.
- 46. Morales AJ, Laughlin GA, Butzow T, Maheshwari H, Baumann G, Yen SS. Insulin, somatotropic, and luteinizing hormone axes in lean and obese women with polycystic ovary syndrome: common and distinct features. J Clin Endocrinol Metab. 1996;81:2854-64.
- 47. Cataldo NA. Insulin-like growth factor binding proteins: do they play a role in polycystic ovary syndrome? Semin Reprod Endocrinol. 1997;15:123-36.
- 48. Barbieri RL, Makris A, Randall RW, Daniels G, Kistner RW, Ryan KJ. Insulin stimulates androgen accumulation in incubations of ovarian stroma obtained from women with hyperandrogenism. J Clin Endocrinol Metab. 1986;62:904-10.
- 49. Tsilchorozidou T, Overton C, Conway GS. The pathophysiologyovary syndrome. Clin Endocrinol. 2004; 60:1-17.
- 50. Escobar-Morreale HF, Roldan B, Barrio R, Alonso M, Sancho J, de la Calle H, et al. High prevalence of the polycystic ovary syndrome and hirsutism in women with type 1 diabetes mellitus. J Clin Endocrinol Metab. 2000;85:4182-7.
- 51. Vollenhoven B, Clark S, Kovacs G, Burger H, Healy D. Prevalence of gestational diabetes mellitus in polycystic ovarian syndrome (PCOS) patients pregnant after ovulation induction with gonadotrophins. Aust N Z J Obstet Gynaecol. 2000;40:54-8.
- 52. Bray GA, York DA. Clinical review 90: leptin and clinical medicine: a new piece in the puzzle of obesity. J Clin Endocrinol Metab. 1997;82: 2771-6.
- 53. Vauhkonen I, Niskanen L, Haffner S, Kainulainen S, Uusitupa M, Laakso M. Insulin resistant phenotype is associated with high serum leptin levels in offspring of patients with non-insulin-dependent diabetes mellitus. Eur J Endocrinol. 1998;139:598-604.
- 54. Karlsson C, Lindell K, Svensson E, Bergh C, Lind P, Billig H, et al. Expression of functional leptin receptors in the human ovary. J Clin Endocrinol Metab. 1997;82:4144-8.
- 55. Jacobs HS, Conway GS. Leptin, polycystic ovaries and polycystic ovary syndrome. Hum Reprod Update. 1999;5:166-71.
- 56. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001; 409: 307-12.
- 57. Talbott E, Clerici A, Berga SL, Kuller L, Guzick D, Detre K, et al. Adverse lipid and coronary heart disease risk profiles in young women with polycystic ovary syndrome: results of a case-control study. J Clin Epidemiol. 1998;51:415-22.
- 58. Marshall JC, Eagleson CA. Neuroendocrine aspects of polycystic ovary syndrome. Endocrinol Metab Clin North Am. 1999;8:295-324.
- 59. Apter D, Butzow T, Laughlin GA, Yen SS. Metabolic features of polycystic ovary syndrome are found in adolescent girls with hyperandrogenism. J Clin Endocrinol Metab. 1995;80:2966-73.
- 60. Lobo RA, Granger L, Goebelsmann U, Mishell DR Jr. Elevations in unbound serum estradiol as a possible mechanism for inappropriate gonadotropin secretion in women with PCO. J Clin Endocrinol Metab.1981;52:156-8.
- 61. Arroyo A, Laughlin GA, Morales AJ, Yen SS. Inappropriate gonadotropin secretion in polycystic ovary syndrome: influence of adiposity. J Clin Endocrinol Metab. 1997;82:3728-33.
- 62. Conway GS, Honour JW, Jacobs HS. Heterogeneity of the polycystic ovary syndrome: clinical, endocrine and ultrasound features in 556 patients. Clin Endocrinol. 1989;30:459-70.
- 63. Ramanujam LN, Liao WX, Roy AC, Loganath A, Goh HH. Association of molecular variants of luteinizing hormone with menstrual disorders. Clin Endocrinol. 1999;51:243-6.
- 64. Liao WX, Roy AC, Chan C, Arulkumaran S, Ratnam SS. A new molecular variant of luteinizing hormone associated with female infertility. Fertil Steril. 1998;69:102-6.
- 65. Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev. 1995;16:322-53.
- 66. Norman RJ, Milner CR, Groome NP, Robertson DM. Circulating follistatin concentrations are higher and activin concentrations are lower in polycystic ovarian syndrome. Hum Reprod. 2001;16:668-72.
- 67. Bleau G, Roberts KD, Chapdelaine A. The in vitro and in vivo uptake and metabolism of steroids in human adipose tissue. J Clin Endocrinol Metab. 1974;39:236-46.
- 68. Stewart PM, Shackleton CH, Beastall GH, Edwards CR. 5 alpha-reductase activity in polycystic ovary syndrome. Lancet. 1990;335:431-3.
- 69. Rodin A, Thakkar H, Taylor N, Clayton R. Hyperandrogenism in polycystic ovary syndrome. Evidence of dysregulation of 11 beta-hydroxysteroid dehydrogenase. N Engl J Med. 1994;330:460-5.
- 70. Ehrhart-Bornstein M, Hinson JP, Bornstein SR, Scherbaum WA, Vinson GP. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocr Rev. 1998;19:101-43.
- 71. Urbanek M, Spielman RS. Genetic analysis of candidate genes for the polycystic ovary syndrome. Curr Opin Endocrinol Diabetes. 2002; 9:492-501.
- 72. Norman RJ, Wu R, Stankiewicz MT. Polycystic ovary syndrome. Med J Aust. 2004;180:132-7.
- 73. Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85:2402-10.
- 74. Diamanti-Kandarakis E, Kouli C, Alexandraki K, Spina G. Failure of mathematical indices to accurately assess insulin resistance in ean, overweight, or obese women with polycystic ovary syndrome. J Clin Endocrinol. Metab. 2004;89:1273-6.
- 75. Legro RS, Finegood D, Dunaif A. A fasting glucose to insulin ratio is a useful measure of insulin sensitivity in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1998; 83:2694-8.
- 76. Sharpless JL. Polycystic ovary syndrome and the metabolic syndrome. Clin Diabets. 2003;21:154-61.
- 77. Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev. 2000;21:363-92.
- 78. Diamanti-Kandarakis E, Baillargeon JP, Iuorno MJ, Jakubowicz DJ, Nestler JE. A modern medical quandary: polycystic ovary syndrome, insulin resistance, and oral contraceptive pills J. Clin. Endocrinol. Metab. 2002;88:1927-32.
- 79. Rittmaster RS. Clinical review 73: medical treatment of androgen-dependent hirsutism. J Clin Endocrinol Metab. 1995;80:2559-63.
- 80. Shaw JC. Low- dose adjunctive spironolactone in treatment of acne in women: A retrospective analysis of 85 consecutively treated patients. J Am Acad Dermatol. 2000;43:498-502.
- 81. Tartagni M, Schonauer LM, De Salvia MA, Cicinelli E, De Pergola G, DAddario V. Comparison of Diane 35 and Diane 35 plus finasteride in the treatment of hirsutism. Fertil Steril. 2000;73:718-23.
- 82. Balfour JA, McClellan K. Topical eflornithine. Am J Clin Dermatol. 2001;2:197-201.
- 83. Velazquez EM, Mendoza S, Hamer T, Sosa F, Glueck CJ. Metformin therapy in polycystic ovary syndrome reduces hyperinsulinemia, insulin resistance, hyperandrogenemia, and systolic blood pressure, while facilitating normal menses and pregnancy. Metab Clin Exp. 1994;43:647-54.
- 84. Mansfield R, Galea R, Brincat M, Hole D, Mason H. Metformin has direct effects on human ovarian steroidogenesis. Fertil Steril. 2003;79:956-62.
- 85. Azziz R, Ehrmann D, Legro RS, Whitcomb RW, Hanley R, Fereshetian AG, et al. Troglitazone improves ovulation and hirsutism in the polycystic ovary syndrome: a multicenter, double blind, placebo-controlled trial. J Clin Endocrinol Metab. 2001;86:1626-32.
- 86. Lord JM, Flight IH, Norman RJ. Insulin-sensitising drugs (metformin, troglitazone, rosiglitazone, pioglitazone, D-chiro-inositol) for polycystic ovary syndrome. Cochrane Database Syst Rev. 2003;3:CD003053.
- 87. Ganie MA, Khurana ML., Eunice M, Gulati M, Dwivedi SN, Ammini AC. Comparison of efficacy of spironolactone with metformin in the management of polycystic ovary syndrome: an open-labeled study. J Clin Endocrinol. Metab. 2004;89:2756-62.
- 88. Ortega-González C, Luna S, Hernández L, Crespo G, Aguayo P, Arteaga-Troncoso G, Parra A. Responses of serum androgen and insulin resistance to metformin and pioglitazone in obese, insulin-resistant women with polycystic ovary syndrome. J Clin Endocrinol. Metab. 2004;90:1360-5.
- 89. Nestler JE, Jakubowicz DJ, Reamer P, Gunn RD, Allan G. Ovulatory and metabolic effects of D-chiro-inositol in the polycystic ovary syndrome. N Engl J Med. 1999;340;1314-20.
Publication Dates
-
Publication in this collection
13 Dec 2006 -
Date of issue
Aug 2005
History
-
Accepted
04 July 2005 -
Received
31 May 2005