Abstracts
Increasing knowledge of genomic DNA sequences and genetic databases has led to the characterization of the molecular basis of several inherited skin disorders. In this review we summarize some of the major recent discoveries that have been made in defining the pathogenic mutations that cause inherited disorders of the skin barrier leading to skin scaling or increased transepidermal water loss in either rare disorders (Netherton’s syndrome or harlequin ichthyosis) or more common genodermatoses (ichthyosis vulgaris). These molecular breakthroughs have led to more accurate diagnoses, better genetic counselling and, where appropriate, the feasibility of DNA-based prenatal diagnosis, as well as the possibility of developing newer forms of treatment, including gene or protein therapy. Identifying the molecular basis of these conditions, especially ichthyosis vulgaris, has also provided dramatic new insight into the genetic abnormalities in the common disorder, atopic dermatitis. Thus research on the relatively rare single gene inherited skin disorders not only has benefits for patients and their families with these uncommon conditions but also has the potential to yield fresh and significant new information about very common skin diseases.
Eczema; Epidermis; Ichthyosis; Mutation
O maior conhecimento sobre as de sequências genômicas de DNA e as bases de dados genéticas levou à caracterização da base molecular de várias doenças hereditárias de pele. Nesta revisão resumimos algumas das descobertas recentes mais importantes quanto à definição das mutações patogênicas que causam as doenças hereditárias da barreira cutânea, levando a descamação ou aumento da perda hídrica transepidérmica, seja em doenças raras, (síndrome de Netherton ou ictiose em Arlequim) ou genodermatoses mais comuns (ictiose vulgar). Estas descobertas moleculares têm conduzido a diagnósticos mais acurados, melhor aconselhamento genético e, quando apropriado, à possibilidade de diagnóstico pré-natal baseado em DNA, assim como desenvolvimento de novas formas de tratamento, incluindo terapia gênica ou proteica. A identificação da base molecular destas doenças, especialmente ictiose vulgar, também propiciou importante entendimento das anormalidades genéticas de doença comum, a dermatite atópica. Assim, as pesquisas em doenças hereditárias monogênicas de pele relativamente raras não apenas trazem benefícios para os pacientes e familiares, mas também têm o potencial de trazer informações novas e significativas sobre doenças de pele muito comuns.
Eczema; Epiderme; Ictiose; Mutação
REVIEW ARTICLE
Advances in understanding the genetic basis of inherited single gene skin barrier disorders: new clues to key genes that may be involved in the pathogenesis of atopic dermatitis* * Work done at St Johns Institute of Dermatology, Division of Genetics and Molecular Medicine, The Guys, Kings and St Thomas Hospitals Medical School, Kings College London, UK
Joey E Lai-Cheong; John A. McGrath
Genetic Skin Disease Group, St Johns Institute of Dermatology, Division of Genetics and Molecular Medicine, The Guys, Kings and St Thomas Hospitals Medical School, Kings College London, UK
Corresponding author Corresponding author: John McGrath, Genetic Skin Disease Group, St Johns Institute of Dermatology, St Thomas Hospital, Lambeth Palace Road, London, UK. e-mail: john.mcgrath@kcl.ac.uk tel : 44-20-71886409 fax : 44-20-71886379
ABSTRACT
Increasing knowledge of genomic DNA sequences and genetic databases has led to the characterization of the molecular basis of several inherited skin disorders. In this review we summarize some of the major recent discoveries that have been made in defining the pathogenic mutations that cause inherited disorders of the skin barrier leading to skin scaling or increased transepidermal water loss in either rare disorders (Nethertons syndrome or harlequin ichthyosis) or more common genodermatoses (ichthyosis vulgaris). These molecular breakthroughs have led to more accurate diagnoses, better genetic counselling and, where appropriate, the feasibility of DNA-based prenatal diagnosis, as well as the possibility of developing newer forms of treatment, including gene or protein therapy. Identifying the molecular basis of these conditions, especially ichthyosis vulgaris, has also provided dramatic new insight into the genetic abnormalities in the common disorder, atopic dermatitis. Thus research on the relatively rare single gene inherited skin disorders not only has benefits for patients and their families with these uncommon conditions but also has the potential to yield fresh and significant new information about very common skin diseases.
Keywords: Eczema; Epidermis; Ichthyosis; Mutation
INTRODUCTION
During the last two decades the molecular basis of over 2000 inherited single gene disorders has been determined. More than 25% of these have a skin phenotype, making the inherited skin disorders one of the best characterized groups of genetic diseases. For clinicians, the accumulating molecular data have several benefits for patients. First, access to DNA sequencing laboratories that can identify pathogenic mutations in a particular gene allows more accurate diagnoses to be made. Moreover, the nature of the mutation(s) may provide additional information about inheritance patterns as well as the likely clinical course and prognosis, collectively improving genetic counselling. Secondly, knowledge of family-specific gene mutations makes it possible to undertake DNA-based prenatal diagnosis in subsequent at risk pregnancies. Examination of fetal DNA during the first trimester following chorionic villus biopsy is now feasible for a broad range of severe genodermatoses. Furthermore, preimplantation genetic diagnosis has become a viable option in certain countries. Finally, a key objective of molecular research on the genodermatoses is to develop new treatments that benefit patients. Gene therapy is a science that is very much in its infancy: there are several technical, practical and safety issues that still need to be developed and optimized, and therefore the next few years are likely to see pilot trials of human gene therapy rather than widespread clinical application of such treatments. In essence, the big recent advances in the genodermatoses have been in the plethora of new information available, the rapid access to comprehensive internet databases, and the technological improvements in disease gene identification and mutation detection. Clinically, at this time-point, the outcomes for patients afflicted with genodermatoses are less dramatic: better diagnosis and counselling, more options for prenatal diagnosis, and the prospect (but not yet realization) of new molecular therapies to come. However, research on rare genodermatoses also has the potential to provide new insight into the pathogenesis and pathophysiology of skin diseases that are more frequently seen in dermatological practice. In this review we focus on the recent genetic discoveries that underlie certain forms of ichthyosis or impaired skin barrier formation. The emerging data have immediate but limited benefits for patients and families with these rare conditions but provide dramatic new insight into the molecular pathology of a much more common skin disorder, atopic dermatitis (AD).
Netherton Syndrome
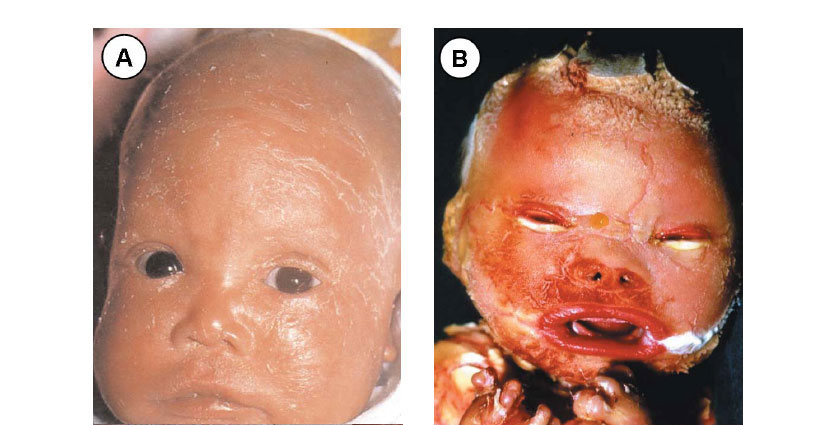
Netherton syndrome (NS) (Figure 1A) is a rare autosomal recessive genodermatosis associated with congenital ichthyosiform erythroderma, hair shaft abnormalities (e.g. trichorrhexis invaginata) and an atopic tendency associated with high circulating IgE levels.1 Identifying the gene for NS, therefore, might be expected to give some clues to the genetic basis of AD. In 2000, the gene for NS was mapped to chromosome 5q32 and was identified as SPINK5, which encodes the serine protease inhibitor LEKTI (lympho-epithelial Kazal-type related inhibitor).2 In NS, loss-offunction mutations in LEKTI lead to increased proteolytic activity in the skin, affecting desquamation and barrier function.3-5 In spink5-null mice, the abnormal proteolysis causes degradation of epidermal desmosomal proteins, particularly desmoglein 1 and desmocollin 1, leading to premature loss of corneodesmosomes.4,5 Moreover, in transgenic mice harbouring a spink5 nonsense mutation (similar to human NS), there is increased processing of the epidermal protein profilaggrin, resulting in an overexpression of filaggrin.6 Filaggrin imparts structural integrity to the stratum corneum but its overexpression can lead to decreased cellular cohesion, possibly through an altered distribution of desmosomal proteins.7,8 These observations clearly indicate an important role for filaggrin in the regulation of epidermal barrier function. Because of the association of atopy and NS, however, genomic DNA from individuals with AD has been examined for mutations or polymorphisms in SPINK5. One particular missense polymorphism, Glu420>Lys, has been shown to be associated with atopy, asthma and AD, especially if maternally inherited. 9 However, the increased risk is only about fourfold and it is evident that sequence alterations in SPINK5 do not account for the major genetic factors relevant to most individuals with AD and therefore other rare diseases will have to be dissected at a molecular level if this candidate approach is to prove more insightful into explaining common skin disease pathophysiology
Harlequin Ichthyosis
A further rare genetic disease that might give clues into key genes that are relevant to AD is the autosomal recessive disorder, harlequin ichthyosis (HI)10 (Figure 1B). This condition is characterized at birth by a thick armour-like plate of scale involving the whole skin leading to impaired thermoregulation, fluid balance and protection against infection. The thick keratin layer also leads to contraction of the skin around the eyes causing ectropion, and the lips resulting in eclabion, and the chest leading to respiratory difficulties. Infants born with HI often die within the first few days of life but some survive with the appropriate clinical care and the early use of retinoids.8,11 In HI there is clearly a grossly abnormal epidermal barrier but it is plausible that less disruptive abnormalities in the gene responsible for HI might also provide some clues to epidermal barrier dysfunction in AD. In 2005, the genetic basis of HI was resolved by two independent groups using single nucleotide polymorphism chip technology with homozygosity mapping or a candidate gene approach.12,13 The gene was mapped to chromosome 2q35 and is known as ABCA12, which encodes a transmembranous regulatory protein involved in lipid transport. Deletion, truncation and some missense mutations result in a dysfunctional protein and lead to HI whilst replacing the wild type allele in cultured null keratinocytes leads to recovery of lamellar granule-mediated lipid secretion.12,14 In contrast to the highly disruptive mutations in ABCA12 that underlie HI, it is possible that less severe functional polymorphisms might be present in some individuals with AD. Such sequence alterations could compromise epidermal barrier integrity, increase transepidermal water loss and, perhaps in combination with other factors, lead to AD. Thus far, however, no specific ABCA12 genotypes have been linked to AD.
Ichthyosis Vulgaris
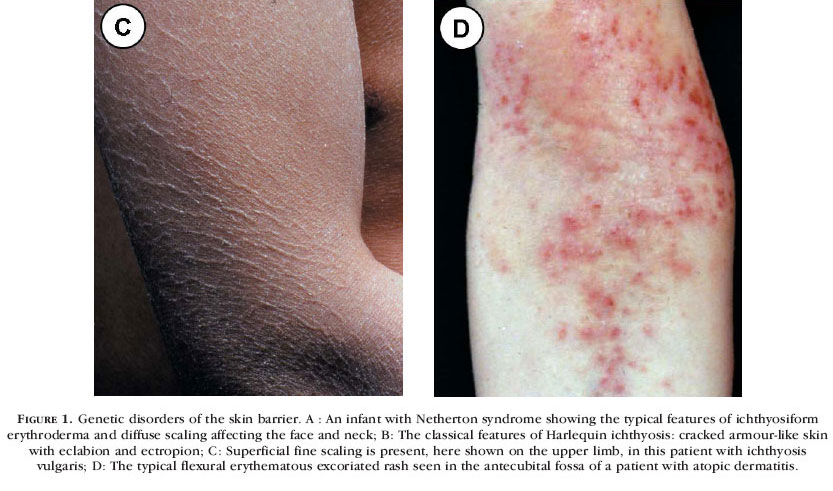
In contrast to the very rare HI, ichthyosis vulgaris (IV) (Figure 1C) is the most common disorder of keratinization, estimated to affect about 1 in 250 people. 15 The clinical features are variable and include keratosis pilaris, fine scale on arms and legs and palmar hyperlinearity, although in some cases the scaling can be extensive and severe. Moreover, the co-existence of IV and AD is well known, with up to 50% of subjects with IV having an atopic tendency,16 and therefore elucidating the molecular basis of IV might be expected to provide new insight into the genetics of AD. Linkage of IV to 1q21 and the epidermal differentiation complex has been previously demonstrated17 but it was not until 2006 that pathogenic mutations were identified in the gene encoding filaggrin (FLG). Notably, homozygous and compound heterozygous mutations including the recurrent mutations R501X and 2282del4 were found in several kindreds with IV. Indeed, these two recurrent mutations were found in about 9% of the European population thereby showing just how common deleterious mutations in FLG actually are. In addition, dominant and semi-dominant mutations in FLG were identified to explain both the differing phenotypic severities of IV and the pattern of inheritance. Notably, a number of mutations were identified that led to ablation of processed filaggrin products.15 Filaggrin is an essential component of the stratum corneum and it is cleaved from profilaggrin causing aggregation of the keratin cytoskeleton during terminal differentiation of the granular layer of the epidermis. Filaggrin is then cross-linked by transglutaminases to contribute to the formation of the cornified cell envelope. In the absence of filaggrin, the epidermal barrier fails to function normally, increasing transepidermal water loss and giving rise to the ichthyosis.
Atopic Dermatitis
Atopic dermatitis (Figure 1D) is a common skin disorder, the aetiology of which is complex and interplay between genetic and environmental factors is believed to be important. The finding of common mutations in FLG in a substantial proportion of the European population led investigators to assess the prevalence of filaggrin pathology in AD. For the mutations R501X and 2282del4, the combined allele frequency in AD is 0.330 compared to 0.042 in the nonatopic population, thus demonstrating just how common these mutations actually are.16 The reason why these discoveries were not realized until 2006 was largely due to technical difficulties experienced by many researchers in amplifying the FLG gene (lots of homologous repeat units with little sequence dissimilarity). Once DNA sequencing became possible, however, the common finding of FLG mutations in AD was a truly remarkable discovery. Of course, loss of filaggrin expression does not provide a full explanation for the genetic basis of AD in all subjects. It is likely that AD is in fact a heterogeneous disorder with some individuals having a primary epidermal barrier defect due to FLG gene mutations, whereas others will have primary immunological abnormalities or alternative primary susceptibilities to inflammation or infection or, most probably, a combination of several genetic factors influencing these skin responses. Interestingly, FLG mutations are also a predisposing factor for asthma, but only when associated with AD rather than clinical asthma as a separate entity.16
understanding of epidermal barrier function. Although an atopic tendency is not a feature of HI, it has been found that the ABCA12 protein transports glucosylceramide which is an important component of the lipid barrier of the stratum corneum.12 Thus replacement of this lipid component in the form of a topical preparation could conceivably be used as treatment for not only HI but perhaps as a supplementary agent for other more common dry skin conditions such as AD. Demonstration of FLG mutations in AD, however, perhaps holds the greatest promise for new treatments, for example involving drugs that influence post-transcriptional control of gene expression, or other strategies that restore filaggrin protein expression in the skin thereby restoring the integrity of the skin barrier. It is very likely that new therapies that restore filaggrin expression and function in the skin will emerge within the next few years that have a substantial benefit for a significant proportion of people with AD.
CONCLUSIONS
The molecular characterization of inherited single gene skin disorders such as HI, NS and IV is important not only for patients and their families but also for our understanding of epidermal physiology and pathology. This has direct implications for more common disorders of epidermal dysfunction such as AD. In the future, systematic analysis of the roles of the various proteins involved such as LEKTI, glucosylceramide and, in particular, filaggrin in the maintenance of epidermal integrity is expected to contribute to the development of novel therapeutic strategies and thus represent a direct benefit of translational research on rare genodermatoses to the management of very common skin problems encountered by dermatologists in daily clinical practice.
No conflict of interests was declared.
- 1. Netherton EW. A unique case of trichorrhexis nodosa; bamboo hairs. AMA Arch Derm. 1958;78:483-7.
- 2. Chavanas S, Garner C, Bodemer C, Ali M, Teillac DH, Wilkinson J, et al. Localization of the Netherton sydrome gene to chromosome 5q32, by linkage analysis and homozygosity mapping. Am J Hum Genet. 2000;66:914-21.
- 3. Sprecher E, Chavanas S, DiGiovanna JJ, Amin S, Nielsen K, Prendiville JS, et al. The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syn drome: implications for mutation detection and first case of prenatal diagnosis. J Invest Dermatol. 2001;117:179-87.
- 4. Descargues P, Deraison C, Bonnart C, Kreft M, Kishibe M, Ishida-Yamamoto A, et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat Genet. 2005;37:56-65.
- 5. Descargues P, Deraison C, Prost C, Fraitag S, Mazereeuw-Hautier J, D'Alessio M, et al. Corneodesmosomal cadherins are preferential targets of stratum corneum trypsin- and chymotrypsin-like hyperactivity in Netherton syndrome. J Invest Dermatol. 2006;126:1622-32.
- 6. Hewett DR, Simons AL, Mangan NE, Jolin HE, Green SM, Fallon PG, et al. Lethal, neonatal ichthyosis with increased proteolytic processing of filaggrin in a mouse model of Netherton syndrome. Hum Mol Genet. 2005;14:335-46.
- 7. Presland RB, Kuechle MK, Lewis SP, Fleckman P, Dale BA. Regulated expression of human filaggrin in ker atinocytes results in cytoskeletal disruption, loss of cell-cell adhesion, and cell cycle arrest. Exp Cell Res. 2001;270:199-213.
- 8. Lawlor F, Peiris S. Harlequin fetus successfully treated with etretinate. Br J Dermatol. 1985;112:585-90.
- 9. Walley AJ, Chavanas S, Moffatt MF, Esnouf RM, Ubhi B, Lawrence R, et al. Gene polymorphism in Netherton and common atopic disease. Nat Genet. 2001;29:175-8.
- 10. Hovnanian A. Harlequin ichthyosis unmasked: a defect of lipid transport. J Clin Invest. 2005;115:1708-10.
- 11. Lawlor F, Peiris S. Progress of a harlequin fetus treated with etretinate. J R Soc Med. 1985;78(Suppl 11):S1920.
- 12. Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777-84.
- 13. Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, et al. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet. 2005;76:794-803.
- 14. Hansson L, Backman A, Ny A, Edlund M, Ekholm E, Ekstrand Hammarstrom B, et al. Epidermal overexpres sion of stratum corneum chymotryptic enzyme in mice: a model for chronic itchy dermatitis. J Invest Dermatol. 2002;118:444-9.
- 15. Smith FJ, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38:337-42.
- 16. Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441-6.
- 17. Mischke D, Korge BP, Marenholz I, Volz A, Ziegler A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex ("epidermal differentiation complex") on human chromosome 1q21. J Invest Dermatol. 1996;106:989-92.
Publication Dates
-
Publication in this collection
21 June 2007 -
Date of issue
Dec 2006