Resumos
Dermatomiosite é doença idiopática inflamatória crônica que afeta a musculatura estriada, a pele e outros órgãos. Apresenta critérios diagnósticos definidos por Bohan & Peter, podendo os pacientes ser classificados em cinco grupos: dermatomiosite juvenil, dermatomiosite primária idiopática, dermatomiosites amiopáticas, dermatomiosite associada a neoplasias e dermatomiosite associada a outras doenças do tecido conectivo. O sexo feminino é mais afetado, e a idade média do diagnóstico é 40 anos. Manifestações cutâneas são observadas em todos os pacientes. Das alterações sistêmicas, a manifestação muscular mais freqüente é a perda de força proximal, e a manifestação pulmonar mais comum é a pneumopatia intersticial. Podem ser observadas neoplasias durante o seguimento da doença, sendo mais freqüentes nos pacientes acima de 60 anos. A desidrogenase lática é a enzima muscular alterada na maioria dos casos. Para diagnóstico da dermatomiosite, pode ser realizado exame anatomopatológico de biópsia cutânea e biópsia muscular, além de eletroneuromiografia. Os corticóides são a terapia mais utilizada. As causas de óbito mais freqüentes são a neoplasia maligna, a septicemia e a infecção pulmonar.
Dermatomiosite; Dermatomiosite; Dermatomiosite; Dermatomiosite; Doenças auto-imunes; Doenças do tecido conjuntivo; Doenças musculares
Dermatomyositis is a chronic idiopathic inflammatory disorder that affects striated skeletal muscles, the skin, and other organs. Diagnostic criteria were established by Bohan & Peter and patients may be classified into five groups: juvenile dermatomyositis, primary dermatomyositis, amyopatic dermatomyositis, dermatomyositis associated with malignancies and dermatomyositis associated with other connective tissue disorders. Females are more affected and the mean age of diagnosis is 40 years. Skin manifestations are observed in all patients. Loss of proximal strength is the most common systemic alteration and lung involvement is most often manifested as interstitial pneumopathy. Neoplasms may be detected during the course of the disease specially in patients over 60. Lactic dehydrogenase serum levels are altered in the majority of cases and diagnosis can be established or the basis of skin and muscle biopsies and electroneuromiography. Corticosteroids are the first line drugs. The most common causes of death are malignant neoplasms, sepsis and pulmonary infection.
Autoimmune diseases; Connective tissue diseases; Dermatomyositis; Dermatomyositis; Dermatomyositis; Dermatomyositis; Muscular diseases
ARTIGO DE REVISÃO
Dermatomiosite* * Trabalho realizado no Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo (FMUSP) São Paulo (SP), Brasil. Conflito de interesse: Nenhum Suporte financeiro: Nenhum
Dermatomyositis* * Trabalho realizado no Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo (FMUSP) São Paulo (SP), Brasil. Conflito de interesse: Nenhum Suporte financeiro: Nenhum
Luciena Cegatto Martins OrtigosaI; Vitor Manoel Silva dos ReisII
IProfessora do Departamento de Dermatologia, Faculdade de Medicina da Universidade do Oeste Paulista (Unoeste), Presidente Prudente (SP). Mestre em Dermatologia pela Universidade de São Paulo (USP) - São Paulo (SP), Brasil
IIProfessor Dr. do Departamento de Dermatologia, Faculdade de Medicina da Universidade de São Paulo (FMUSP) - São Paulo (SP), Brasil
Endereço para correspondência/Mailing Address Endereço para correspondência/Mailing Address: Luciena Cegatto Martins Ortigosa Rua Doze de outubro, 1696 - Centro. 19015 090 - Presidente Prudente - SP Tel./Fax: 11 - 39163642 / 39081823 E-mail: luciena@speedymed.com.br
RESUMO
Dermatomiosite é doença idiopática inflamatória crônica que afeta a musculatura estriada, a pele e outros órgãos. Apresenta critérios diagnósticos definidos por Bohan & Peter, podendo os pacientes ser classificados em cinco grupos: dermatomiosite juvenil, dermatomiosite primária idiopática, dermatomiosites amiopáticas, dermatomiosite associada a neoplasias e dermatomiosite associada a outras doenças do tecido conectivo. O sexo feminino é mais afetado, e a idade média do diagnóstico é 40 anos. Manifestações cutâneas são observadas em todos os pacientes. Das alterações sistêmicas, a manifestação muscular mais freqüente é a perda de força proximal, e a manifestação pulmonar mais comum é a pneumopatia intersticial. Podem ser observadas neoplasias durante o seguimento da doença, sendo mais freqüentes nos pacientes acima de 60 anos. A desidrogenase lática é a enzima muscular alterada na maioria dos casos. Para diagnóstico da dermatomiosite, pode ser realizado exame anatomopatológico de biópsia cutânea e biópsia muscular, além de eletroneuromiografia. Os corticóides são a terapia mais utilizada. As causas de óbito mais freqüentes são a neoplasia maligna, a septicemia e a infecção pulmonar.
Palavras-chave: Dermatomiosite/classificação; Dermatomiosite/diagnóstico; Dermatomiosite/etiologia; Dermatomiosite/terapia; Doenças auto-imunes; Doenças do tecido conjuntivo; Doenças musculares
ABSTRACT
Dermatomyositis is a chronic idiopathic inflammatory disorder that affects striated skeletal muscles, the skin, and other organs. Diagnostic criteria were established by Bohan & Peter and patients may be classified into five groups: juvenile dermatomyositis, primary dermatomyositis, amyopatic dermatomyositis, dermatomyositis associated with malignancies and dermatomyositis associated with other connective tissue disorders. Females are more affected and the mean age of diagnosis is 40 years. Skin manifestations are observed in all patients. Loss of proximal strength is the most common systemic alteration and lung involvement is most often manifested as interstitial pneumopathy. Neoplasms may be detected during the course of the disease specially in patients over 60. Lactic dehydrogenase serum levels are altered in the majority of cases and diagnosis can be established or the basis of skin and muscle biopsies and electroneuromiography. Corticosteroids are the first line drugs. The most common causes of death are malignant neoplasms, sepsis and pulmonary infection.
Keywords: Autoimmune diseases; Connective tissue diseases; Dermatomyositis/classification;Dermatomyositis/diagnosis; Dermatomyositis/etiology; Dermatomyositis/therapy; Muscular diseases
INTRODUÇÃO
A dermatomiosite (DM) é doença do tecido conjuntivo que associa miopatia a manifestações cutâneas características, cuja causa permanece desconhecida, sendo considerada doença idiopática.1,2 Na etiologia, consideram-se as associações com antígenos de histocompatibilidade, vírus, drogas e auto-imunidade.
Foi reconhecida há mais de 100 anos 3 e descrita na literatura por Wagner.4 Denominada inicialmente pseudotriquinose, foi associada a reação do sistema retículo-endotelial.5 A associação com neoplasia foi publicada pela primeira vez por Stertz em 1916.6, 7
A classificação foi proposta por Bohan e Peter 8 e revista, em 1996, por Drake.9
Pacientes com DM apresentam manifestações cutâneas e sistêmicas, sendo mais comuns lesões em áreas fotoexpostas, fraqueza muscular proximal, alterações da musculatura respiratória e disfagia.2 Na forma juvenil, há maior incidência de calcinose cutânea.10,11
O diagnóstico é realizado através da história e exame físico, sendo corroborado pelas enzimas musculares, biópsia muscular, biópsia cutânea, eletroneuromiografia (ENMG), ultra-sonografia (USG) e ressonância nuclear magnética (RNM).2,10
O interesse pela dermatomiosite vem aumentando na última década não apenas pelo maior conhecimento imunológico sobre a doença, mas também devido ao progresso das técnicas de imagem, que tanto auxiliam no diagnóstico.
DEFINIÇÃO E CRITÉRIO
As miopatias inflamatórias idiopáticas (Quadro 1) têm como característica histopatológica o processo inflamatório em músculos estriados.12 Alterações cutâneas são encontradas tanto na DM como na miosite por corpos de inclusão.3,13-15
Bohan e Peter classificaram a DM em DM primária idiopática (DMPI), DM juvenil (DMJ), DM associada a neoplasia e DM associada a outra doença do tecido conjuntivo (DMDC). Definiram, também, critérios para seu diagnóstico (Quadro 2). A doença é considerada "possível" quando existem dois dos cinco critérios, "provável" com três e "definida" com a presença de quatro critérios.5,12 Em 1996, Drake acrescentou a DM amiopática (DMA) a essa classificação.9
EPIDEMIOLOGIA
A DM tem incidência de dois a sete casos novos por milhão de habitantes/ano 3,16,17 e prevalência de 10 a 60 casos por milhão de habitantes/ano.5 Apresenta dois picos de incidência: cinco a 14 anos 2 e 45 a 65 anos.14,18 A idade do diagnóstico é, em média, 40 anos. Na DMJ a incidência é de um a três casos novos por milhão de crianças/ano.2,5 É excepcionalmente encontrada no recém-nascido.5,18
A DMA ocorre em 10% dos casos e é mais comum em adultos.9 A DM é mais comum em mulheres (2:1), com aumento dessas proporções em DMDC.15,17,18 Na DMN e na DMJ essa relação se repete.3
ETIOLOGIA
A DM é relacionada à auto-imunidade, sendo presentes os antígenos de histocompatibilidade (HLA): HLA-B8, HLA-B14, HLA-DR3, HLA-DRw52 e HLA-DQA1. Quando induzida por drogas, está associada a HLA-B18, HLA-B35 e HLA-DR414 e na DMJ a HLAB8, 3,19 HLA-DR3 e HLA-DQA1*0501.18 A DMDC está relacionada a HLA B14, B40,3 B7 e DRB1*0101.19 A associação da doença com HLA é tão importante, que Medsger e Oddis 20 propõem a subclassificação da DM em dois grandes grupos imunogenéticos: HLA DRw52 (miosite mais grave e pior prognóstico) e HLA DRw53 (miosite menos grave e com melhor prognóstico). A DM familiar é rara, tendo sido descritos alguns casos, principalmente na DMJ.21
Infecções virais são precipitantes da DM, tais como influenza A, hepatite B, Coxsackie e Picornavírus. A miosite ocorre também após infecção por Echovírus, HIV e HTLV.3,22 Apesar dessas associações, estudo realizado por Pachman et al.23 não demonstrou presença de RNA viral em músculos de crianças com DM. Há ainda associação com protozoários, como o Toxoplama gondii.5
As lesões cutâneas e musculares são agravadas pelo sol.24 Fototestes, entretanto, não reproduzem as lesões cutâneas, sendo o espectro de luz que causa as manifestações clínicas ainda desconhecido.25 Segundo Hengstman et al.,26 uma das prováveis causas da DM é a radiação ultravioleta B.
Algumas drogas são precipitantes da doença.3 Há suspeita de associação com implante de silicone.27,28
A DM resulta de processo mediado por alterações imunológicas iniciadas por fatores como malignidade, drogas e agentes infecciosos em indivíduos geneticamente predispostos.19 A associação a outras doenças auto-imunes demonstra a essência da origem auto-imune da doença.3
PATOGENIA
A DM envolve microvasculopatia mediada por complemento, apresentando depósitos de complexos C5b-9 de ataque às membranas na junção dermoepidérmica e em vasos da derme.5,29
O mecanismo humoral da DM é a base da microangiopatia muscular, com células CD4, células B, depósitos de imunoglobulinas e complemento, diferindo da polimiosite (PM), em que há atividade de células CD8 contra antígeno muscular específico.15 Complexos C5b-9 nas arteríolas musculares são os causadores dos microtrombos com infartos a distância. As alterações capilares precedem outras alterações das fibras musculares, estando o endotélio capilar no centro da resposta auto-imune. Há ativação endotelial pelas citocinas pró-inflamatórias que transformam o potencial antitrombótico em pró-trombótico.18
A imunidade celular também é ativada, principalmente através da apoptose.3,5
No infiltrado inflamatório dos músculos estão presentes a interleucina 1 (IL-1) e o fator de necrose tumoral (TNF) α. A IL-1á presente nas células endoteliais capilares indica a lesão vascular primária como patogênese da doença.30 Nyberg et al.31 demonstraram forte expressão de IL-1 α nos capilares e de MHC-I nas fibras musculares. A ativação de células endoteliais evidencia-se pela presença de moléculas de adesão intercelular 1 (Icam-1) e moléculas de adesão de células vasculares 1 (VCAM-1).18,30 Há relação entre decréscimo da função muscular com aumento da expressão de IL-1 α, moléculas de adesão nas células endoteliais e de MHC-I nas fibras musculares.30
A radiação ultravioleta induz apoptose e translocação de auto-antígenos com formação de bolhas nas membranas plasmáticas e exposição de corpos apoptóticos ao sistema imune, causando respostas mediadas por células T e anticorpos. O mediador desse processo é o TNF α.32
MANIFESTAÇÕES CLÍNICAS
Dermatomiosite primária idiopática
As lesões cutâneas precedem a fraqueza muscular em 56% dos casos; a fraqueza muscular precede as lesões cutâneas em 16% dos casos, e os dois sinais aparecem simultaneamente em 28% dos pacientes.3, 14
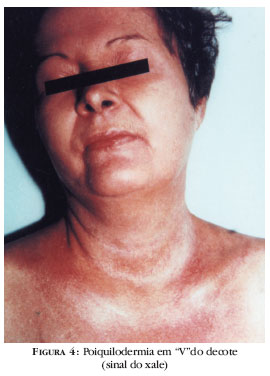
Alterações da pele podem ser divididas em patognomônicas, características e compatíveis.15 As patognomônicas ocorrem em 70% dos casos, com as pápulas de Gottron, que são lesões violáceas sobre articulações interfalangeanas ou metacarpofalangeanas, cotovelos ou joelhos (Figura 1), e o sinal de Gottron, que consiste em eritema violáceo sobre placa atrófica nas mesmas distribuições (Figura 2).2, 15 Lesões características incluem as telangectasias periungueais e distrofias cuticulares,3,15 a mácula eritêmato- vinhosa e edematosa na região periorbital conhecida como heliotropo pela semelhança com a coloração violácea da flor heliotropo3 (Figura 3) e as máculas eritêmato-poiquilodérmicas distribuídas nos ombros, braços, V do decote ou dorso, conhecidas como sinal do xale (Figura 4). O heliotropo ocorre em 30 a 60% dos pacientes, pode envolver apenas a região de pálpebras superiores e, em afro-americanos, apresentar-se como edema periorbitário.2,5 O eritema periungueal (sinal de Keinig)18 e as telangectasias periungueais não são específicos, podendo ocorrer em outras doenças. Lesões compatíveis incluem a poiquilodermia vascular atrofiante (poiquilodermatomiosite) e a calcinose.15
Fotossensibilidade ocorre em um terço dos casos.5,33 Em 82% dos pacientes observa-se envolvimento do couro cabeludo,13 que pode ser confundido com dermatite seborréica ou psoríase, e em alguns casos ocorre alopecia.9,15,17 As "mãos de mecânico" são caracterizadas por descamação, fissuras, ceratose e hiperpigmentação simétricas e não pruriginosas nas palmas.15,34 Outros achados incluem edema facial, ictiose adquirida, vasculite, paniculite, perfuração de septo nasal, eritrodermia, líquen plano, doença de Degos, formação de vésico-bolhas,5 erupção pustulosa de cotovelos e joelhos, telangiectasias gengivais,3 eritema centrífugo flagelado (lesões raras, com eritema linear semelhante a listras de zebra),35 dermatite herpetiforme, penfigóide bolhoso13 e urticária.25
Quadro raro é a variante "Wong" da DM, que apresenta hiperceratose folicular simulando pitiríase rubra pilar, com destruição folicular e miosite de músculos eretores do pêlo ao exame anatomopatológico. 13,36 Também são incomuns malacoplaquia, mucinose, 5,37 lipodistrofia, esclerodermia cutânea localizada 38 e o envolvimento vulvar ou escrotal.2 Prurido é sintoma comum.5,13,15 O envolvimento da pele geralmente melhora com o tratamento, porém não reflete a gravidade da doença ou associação com miopatia, podendo até progredir quando as lesões musculares já estiverem sendo atenuadas.3,5
O quadro muscular inclui a perda de força proximal, principalmente em tríceps e quadríceps,3 ocorrendo de forma simétrica e acentuando-se progressivamente, em geral com caráter subagudo. Disfagia, disfonia e fadiga são sintomas comuns secundários ao envolvimento dos músculos faríngeo e flexor da nuca.2,5 Mialgias ocorrem em mais da metade dos pacientes; a minoria refere artralgias, e a artrite é geralmente não erosiva, não deformante e sem sinovite.3,5
Os reflexos tendíneos não se modificam, e atrofia muscular e lordose ocorrem nas fases tardias da doença. As musculaturas facial e bulbar são poupadas, 18 exceto na associação com neoplasia.14 A disfagia ocorre em 15 a 50% dos pacientes, podendo ser distal ou proximal. Na proximal, há envolvimento dos músculos estriados da faringe ou da musculatura proximal do esôfago, podendo ser sinal de mau prognóstico,15 com associação de pneumopatia intersticial.25 Disfagia distal ocorre quando há envolvimento de músculos não estriados, sendo mais comum em síndromes de superposição.25
Além da pneumopatia intersticial, há complicações pulmonares decorrentes da doença muscular (aspiração e hipoventilação) 25 ou secundárias ao tratamento (reações a drogas, como a pneumonite por metotrexato, e infecções oportunistas).25,39 Podem ocorrer derrame pleural, hipertensão pulmonar, pneumotórax, proteinose alveolar pulmonar 5 e fibrose.14 Alterações pulmonares ocorrem em 15 a 30% dos casos e são fatores de mau prognóstico.3,25 A doença pulmonar intersticial é uma das complicações mais graves, sendo o nível aumentado de proteína D do surfactante na corrente sangüínea marcador da doença.40 Ela ocorre principalmente em pacientes com anticorpos anti-sintetases, entre 40 e 60% dos casos de DM.5
No aparelho cardiovascular, podem ocorrer arritmias, insuficiência cardíaca, pericardite, derrame, tamponamento pericárdico,5,39 miocardite e endomiocardiofibrose. 14 Alterações cardíacas ocorrem em 40 a 50% dos pacientes e também são fatores de mau prognóstico. 13,25
No sistema gastrointestinal são comuns o refluxo esofágico, disfagia, dismotilidade, alterações no peristaltismo e incontinência fecal.5 Complicações incluem perfuração, hemorragia, pseudoobstrução e pneumatose cística intestinal.41
Manifestações oculares incluem edema conjuntival, nistagmo, alterações nos músculos extra-oculares, irite, episclerite, conjuntivite, uveíte, glaucoma, atrofia do nervo óptico e pseudopolipose conjuntival. A retinopatia é rara e pode ser vista na DMJ.5,39,42
Os pacientes com DM apresentam algumas condições que os predispõem a maior risco de infecções e incluem envolvimento esofágico, miosite de musculatura torácica, calcinose cutânea e uso de agentes imunossupressores, resultando em grande porcentagem de óbitos.43
A avaliação de exames subsidiários dos pacientes com DM referente aos vários sistemas deve ser direcionada por seus sintomas.
Dermatomiosite juvenil
É, por definição, a DM que ocorre em indivíduos com menos de 18 anos de idade (Figura 5).4,44-46 A forma juvenil da doença difere da forma adulta por aspectos clínicos e, também, histopatológicos.
As manifestações cutâneas são semelhantes, porém na DMJ há maior incidência de calcinose cutânea, presente entre 30 e 70% nas crianças, dependendo da duração e atividade da doença, e em menos de 10% nos adultos.2,5,17 A calcinose pode estar associada à resistência a corticóides, sendo possível a fistulização para a pele.3
Vasculites sistêmicas são mais freqüentes e podem evoluir com encefalopatia.3,9,15,46,47 Na DMJ, a presença de hipertricose, lipoatrofia 5 e telangiectasias gengivais 48 pode auxiliar no diagnóstico. Febre e artrite simétrica de grandes e pequenas articulações são manifestações freqüentes, ocorrendo contraturas.5
Manifestações gastrointestinais incluem má absorção, alteração na motilidade esofágica, infarto, úlcera, perfuração, hemorragias intestinais 5 e colestase.49
Alterações eletrocardiográficas são vistas em metade dos pacientes e insuficiência ventilatória subclínica na maioria.5
A relação entre DMJ e malignidade é pequena.13 Não se conhece a prevalência da DMA em paciente pediátricos, sendo necessária dosagem das enzimas musculares ou RNM a cada três meses para permitir diagnóstico precoce e tratamento rápido e eficaz.50
Dermatomiosite amiopática
É também denominada DM sem miosite, termo usado para descrever casos com lesões cutâneas típicas e patognomônicas, sem sintomas ou alterações laboratoriais que evidenciem envolvimento dos músculos. 2 A DMA é mais freqüente em adultos e mais incidente em mulheres.15 É entidade ainda controversa, sendo considerada DM improvável se usado o critério de Bohan e Peter.8
Os pacientes queixam-se de letargia, fadiga, prurido, fotossensibilidade e artralgias. Alguns não apresentam miosite clínica nem laboratorial, outros têm alterações clínicas sem alterações laboratoriais, e outros, ainda, alterações laboratoriais sutis, sem sintomas clínicos.5 Manifestações sistêmicas são incomuns, porém pneumonite intersticial e pneumomediastino já foram descritos.13
Mais de um terço dos pacientes com DMPI têm como quadro inicial a lesão cutânea,16 que pode preceder, em anos, as alterações sistêmicas da doença; existindo sua suspeita, a introdução de corticóides pode impedir o surgimento da miosite.5 Quando as lesões cutâneas precedem a miosite, o intervalo para aparecimento de quadro muscular é, em média, de três a seis meses, porém pode demorar até quatro anos.15 Acompanhamento do paciente por no mínimo dois anos é necessário para confirmar o diagnóstico. Entretanto, após seis meses do início dos sintomas sem quadro muscular, já se pode fazer o diagnóstico provisório de DMA.15,16
Os exames necessários para avaliação da DMA incluem as enzimas musculares, a ENMG e a biópsia muscular que, muitas vezes, apresentam algum tipo de alteração, mas sem correlação clínica com a doença. 5 Apesar de biópsia muscular prévia normal, alguns pacientes podem apresentar sinais inflamatórios à RNM e à espectroscopia por RNM, exames utilizados para diagnóstico de lesões musculares mínimas,51 sendo atualmente considerados com DM hipomiopática. 13 No caso da DMA, o diagnóstico baseia-se nos aspectos clínicos característicos, na histologia cutânea e, eventualmente, na capilaroscopia.22
Dermatomiosite associada à doença do tecido conjuntivo
A DMDC (síndrome de superposição) é mais freqüente em afro-americanos e em mulheres, na proporção de 9:1, segundo Kovacs e Kovacs,5 e 3:1, segundo Dourmishev et al. 13.
Queixas como mialgias e fraqueza muscular são comuns na maioria das doenças do tecido conjuntivo e não devem ser confundidas com a DM. Para se fazer o diagnóstico, são necessários os exames com alterações características de miosite.5
As doenças do tecido conjuntivo que podem associar-se à DM são doença mista do colágeno, esclerodermia, lúpus eritematoso, artrite reumatóide, síndrome de Sjögren e poliarterite nodosa. Os pacientes que estiverem nessa categoria devem preencher critérios diagnósticos para as duas doenças separadamente.5,8 A maioria dos que apresentam síndrome de superposição tem esclerodermia sobre DM.15 Pode ocorrer artrite, fenômeno de Raynaud e esclerodactilia.5,13
Forma distinta de DMDC é a esclerodermatomiosite, associação de DM e esclerose sistêmica com curso crônico, relativamente benigno e presença de anticorpos anti-PM-Scl. Todos esses pacientes apresentam HLA-DQAI*050I, e 94% deles, HLA-DRBI*030I.13
Dermatomiosite associada a neoplasia
A doença, quando associada a neoplasias, pode ter início mais agudo, com manifestações cutâneas precoces e intensas, e presença de necrose. As alterações musculares podem ser tardias, porém com gravidade rapidamente progressiva e resistência a corticóides. 6,22
A DMN varia entre menos de 10% e mais de 50% dos casos de DM, dependendo do estudo.3 A relação com neoplasia é mais comum na DM (15%) do que na PM (9%),11,15,52 sendo mais freqüente em adultos. 53 Apesar de o risco de malignidade em idosos independer da presença de miosite, há alta probabilidade de pacientes acima de 45 anos de idade com DM apresentarem algum tipo de neoplasia 5,10,54 que, porém, é mais comum em adultos com mais de 60 anos.17
A gravidade da miopatia inflamatória não está associada à ocorrência de malignidade.12 Dentre as neoplasias relacionadas à DM destacam-se, nas mulheres, 5,11,55-57 o câncer de ovário e, nos homens,5 o de estômago e linfoma, embora se encontrem também outros, como de mama e pulmão,3 neoplasias esofágicas, 58 pancreáticas, colorretais, linfoma não Hodgkin,12 câncer de testículo, próstata,25,59 pênis,60 melanoma, timoma, neoplasias uterinas, sarcoma de Kaposi, meningioma, leucemia, neoplasias renais e do trato geniturinário 14,61-64 e mieloma.65 Peculiaridades regionais, como o carcinoma de nasofaringe em Singapura, também foram relatadas.15,66 Carcinomas são mais freqüentes do que sarcomas e malignidades linfoproliferativas. 15
O diagnóstico da neoplasia pode anteceder a DM (40%), ser subseqüente a ela (34%), ou as duas doenças podem ser diagnosticadas simultaneamente (26%).5,12,14,25 A miosite pode melhorar com o tratamento da neoplasia curso paraneoplásico ou seguir curso próprio.25 O risco de malignidade diminui com o passar dos anos após o diagnóstico, sendo a DMN menos responsiva aos corticóides sistêmicos.15
Acredita-se na possibilidade de o uso de imunossupressores aumentar o risco de câncer. Entretanto, estudos mostram que não existe aumento na prevalência de malignidade nesses pacientes.11,54 O mecanismo de associação entre DM e câncer ainda é desconhecido, sendo possivelmente desencadeado por presença de fatores virais, que funcionam como carcinógenos e indutores de processos inflamatórios.67
O prognóstico da DM é afetado pela associação com neoplasia, a qual aumenta a mortalidade68 e a morbidade da doença.69
Todo paciente suspeito de DMN deve ser devidamente investigado, selecionando-se os exames a serem executados de acordo com sexo e idade, que devem ser repetidos a cada novo sintoma ou, anualmente, nos primeiros três anos após o diagnóstico.25, 56
DIAGNÓSTICO DIFERENCIAL
Em pacientes com suspeita de DM são importantes as avaliações da história clínica, exames físico e laboratoriais, além de ENMG e biópsia muscular. Todas as enzimas musculares devem ser pesquisadas, pois há casos em que apenas uma enzima se encontra alterada,5 sendo mais freqüente o aumento da CK e da aldolase.3
Os diagnósticos diferenciais incluem doenças como erupção polimorfa à luz, lúpus eritematoso sistêmico (LES), líquen plano, linfoma cutâneo,3 dermatite de contato, dermatite seborréica, psoríase, dermatite atópica, rosácea,25 triquinose, infecção por HIV e doença de Lyme.15
O rash do LES é mais avermelhado em contraste com o violáceo da DM e atinge mais as áreas malares do que as periorbitais. As lesões no dorso dos dedos são interarticulares, ao contrário das pápulas de Gottron, que ocorrem justo acima das articulações, além de a miosite no LES não ser tão grave.5
Algumas drogas podem causar DM, tais como penicilamina, antiinflamatórios não esteróides, practolol, hidroxiuréia,5 corticóides, álcool, zidovudina,3 ipeca, antimaláricos,15 antimicrobianos, vacinas, clofibrato, pravastatina13 e triptofano.14 O quadro inicia-se em poucos meses após a introdução do medicamento, exceto com a penicilamina, que pode ocorrer após anos de tratamento. A doença responde bem a corticóides, e a remoção da droga causadora pode não causar a reversão dos sintomas. Existe descrição de alergia a cloreto de benzalcônio, usado como conservante de colírios, simulando o heliotropo.70
Outras doenças podem ser semelhantes à DM, como infecções e infestações (esquistossomose, tripanossomíase, toxoplasmose, estafilococcia),8 alterações endócrinas (hipotireoidismo, hipertireoidismo e doença de Cushing), doenças neurológicas (miastenia grave, esclerose lateral amiotrófica e síndrome de Guillain-Barré), doenças metabólicas (hipocalemia, hipocalcemia e hipofosfatemia),5 além de miosite pós-infecção viral, pós-vacinação anti-rubéola, retículo- histiocitose multicêntrica, microêmbolo ateromatoso e embolização por carcinomas com necrose muscular.8
EXAMES SUBSIDIÁRIOS
Enzimas musculares
São elas a creatinofosfoquinase (CK), alaninoaminotransferase (ALT), aspartatoaminotransferase (AST), desidrogenase lática (DHL) e aldolase. A CK é a mais sensível para o diagnóstico de DM,8,14,33 podendo elevar-se após exercício físico e trauma muscular, não sendo por isso considerada enzima específica para avaliação de miosite. Apesar disso, a CK é usada para monitorização da atividade da doença, pois seus valores se alteram com o grau de lesão muscular. Ela pode estar normal no início da doença ou após grandes atrofias musculares, permanecendo inalterada em alguns pacientes durante todo o curso da DM.5,12 Há aumento dos níveis de CK em alguma fase da doença em 95% dos pacientes.3
ALT e AST não são sensíveis para a avaliação de miosite ou para analisar resposta ao tratamento, mas seus níveis podem estar elevados em casos em que a CK se apresenta normal. A anidrase carbônica III é mais específica para avaliação de miosite do que o DHL e as transaminases, porém seu uso não é amplamente difundido.5
As doenças de neurônios motores, a distrofia muscular de Duchenne, desordens metabólicas, endocrinopatias, toxinas e infecções também podem acompanhar- se de aumento dessas enzimas.8
Auto-anticorpos
A causa da produção de auto-anticorpos permanece desconhecida. Sabe-se que a clivagem de antígenos durante a apoptose celular, principalmente por proteases como a granzima B, produz fragmentos únicos de antígenos com potencial para a formação de auto-anticorpos.71
Eles são classificados em específicos e não específicos para miosite. Entre os não específicos, o fator antinuclear está positivo na maioria dos pacientes,5,9 sendo os títulos mais altos nos casos de DMDC.5,16
O anticorpo anti-RNP está presente quando doença mista do tecido conjuntivo ou LE está relacionado à DM, e o antiPM-Scl quando é a esclerodermia a doença associada. Anticorpos antiKu podem acompanhar DM associada à esclerodermia ou ao LE.5,9
Anticorpos miosite-específicos (MSAs) são encontrados em aproximadamente um terço dos casos 5 e ocorrem, com maior freqüência, não associados à malignidade. Esses anticorpos são direcionados a proteínas citoplasmáticas e a ácidos ribonucléicos envolvidos no processo de síntese protéica. É rara a expressão de mais de um tipo de MSA, 29 sendo que geralmente precedem o aparecimento da miosite, podendo se correlacionar com atividade da doença, desaparecendo após a remissão.5 Há quatro grupos principais de MSAs72 (Quadro 3).
A associação de DM com artrite simétrica não erosiva, pneumopatia intersticial, fenômeno de Raynaud, "mãos de mecânico", febre de origem indeterminada e anticorpos anti-sintetase é chamada de síndrome anti-sintetase. Esses pacientes apresentam miosite aguda e grave, principalmente na primavera, com freqüente associação com HLA-DR3, HLA-DRw52 e HLA-DQAr*050I.13 Anticorpos antiJo-1 são preditivos de doença intersticial pulmonar, porém raramente são vistos na DM, sendo mais freqüentes na PM.9,25 Todos os anticorpos anti-sintetases descritos têm associações clínicas similares, incluindo a já bem conhecida e estabelecida associação de miosite com doença pulmonar intersticial.71
O auto-anticorpo antiMJ é associado à DMJ. Na DMA não há aumento na prevalência de anticorpos específicos ou não específicos.71
Os anticorpos miosite-específicos podem trazer informações adicionais sobre classificação e prognóstico da doença.25
Eletroneuromiografia
Exame importante para diferenciação de processos de origem neural ou muscular. Em músculos comprometidos pela DM há formação de unidades motoras polifásicas de pequena amplitude, potenciais polifásicos de curta duração, descargas repetitivas e bizarras de alta freqüência, ondas positivas pontiagudas e fibrilações musculares.5
Alguns pacientes apresentam alterações compatíveis com neuropatia à ENMG, e, quando isso ocorre no início da doença, sugere-se associação com neoplasia.8,14
A ENMG é teste sensível, porém não específico para DM. A combinação de alterações na ENMG e lesões cutâneas típicas permite o diagnóstico de DM.3,8
Biópsia muscular
É realizada em músculos clinicamente afetados, em geral a musculatura proximal, 5 sempre dando preferência para o músculo tríceps, pois o deltóide é afetado mais tardiamente.3 A histopatologia demonstra alterações características que são as combinações de atrofia de fibras musculares, principalmente do tipo II e perifasciculares, além de necrose, regeneração e hipertrofia de células musculares.18
Notou-se, em biópsias musculares, a diminuição do número de linfócitos nos pacientes com DM. A linfopenia tem origem desconhecida e pode associarse ao aumento da prevalência de neoplasia nesses pacientes.73
Na DMJ pode ocorrer vasculite necrotizante, e na DMN há maior processo inflamatório e fagocitose, devidos à necrose muscular agressiva.8
A eletromicroscopia do músculo inclui anormalidades, como degeneração focal das miofibrilas, formação de massas citoplasmáticas, desorganização dos sarcômeros, ruptura das linhas Z, desorganização nos filamentos de actina-miosina, aumento da formação de vacúolos autofagocíticos e anormalidades mitocondriais. 8
A biópsia muscular é o exame mais importante para o diagnóstico de DM e para a exclusão de outras miopatias inflamatórias, como miosite por corpos de inclusão, infecções parasitárias, toxoplasmose e sarcoidose. 14 Pode estar normal em 10 a 15% dos casos.8 Por ser considerada exame invasivo, alguns médicos relutam em realizá-la, comprometendo, com freqüência, o diagnóstico da doença.
Ressonância nuclear magnética
É usada para guiar a biópsia muscular e para diagnosticar a doença nos casos em que as enzimas musculares são normais.
As alterações incluem edema subcutâneo e nos músculos, depósito de cálcio, infiltração gordurosa e atrofia dos músculos afetados.5 Existe um tipo de imagem altamente sensível e específico o aumento dos sinais nos cortes realizados em T2 na RNM que permite a avaliação das estruturas musculares mais profundas, como os grupos musculares paraespinais.15
Na DMA, solicita-se a RNM para confirmação diagnóstica 5 e, também, em busca de calcinose nos tecidos moles.3 Pode-se realizar, no lugar da RNM, a USG muscular, porém com poucos resultados, comparando- se os dois métodos.3 A espectroscopia por RNM tem-se mostrado mais sensível para detecção de envolvimento da musculatura na DM.14 Park et al.74 realizaram espectroscopia por RNM com P-31 em pacientes com DMA e demonstraram deficiência metabólica apenas após exercícios físicos, diferente dos casos de DMPI, que apresentam essas alterações já em repouso.
Biópsia cutânea
O exame histopatológico das lesões cutâneas é semelhante ao do LE, com hiperqueratose, acantose e atrofia epidérmica, dermatite de interface, infiltrado perivascular de linfócitos, edema dérmico e depósito de mucina (Figura 6). Se inflamação intensa ocorrer, pode-se encontrar depósito fibrinóide subepidérmico. 3,9,14,16 A imuno-histoquímica apresenta aumento da proporção de linfócitos CD4+ e macrófagos HLADR+. 5 Na IFD, porém, depósitos de anticorpos na junção dermoepidérmica, como vistos no LE, não ocorrem com freqüência.29
As pápulas de Gottron mostram acantose e atrofia epidérmica, sendo diagnóstico diferencial da neurodermite. 3 A vasculite leucocitoclástica pode apontar neoplasia associada.75
É considerada exame não específico, sendo útil nos casos em que a DM pode confundir-se com outras doenças, como dermatite de contato fotoalérgica, linfomas cutâneos e erupção polimorfa à luz.9
TRATAMENTO
Trabalhos recentes demonstram tratamento efetivo da doença,76-86 apresentado no quadro 4.
PROGNÓSTICO
Antes do tratamento com prednisona, a mortalidade era de 50% dos casos. Atualmente o índice de mortalidade caiu significativamente.3
Indicadores de mau prognóstico incluem doença recalcitrante, demora no diagnóstico e início do tratamento, idade avançada, associação com malignidade, febre, astenia, anorexia, doença pulmonar intersticial, disfagia e leucocitose. A determinação dos MSAs pode avaliar o prognóstico relacionado à resposta a corticóides, tendo os pacientes com anti- SRP má resposta, os com anti-sintetase, resposta moderada, e aqueles com antiMi-2, boa resposta ao tratamento.68,69,87
Na DMJ, consideram-se indicadores de mau prognóstico o tratamento inicial com baixas doses de corticóide, a demora no início do tratamento, a doença recalcitrante e o envolvimento de músculos faríngeos. 3□
AGRADECIMENTOS
À Dra Neusa Sakai Valente pela fotografia do exame anatomopatológico.
Aprovado pelo Conselho Editorial e aceito para publicação em 29.05.2008.
- 1. Kimball AB, Summers RM, Turner M, Dugan EM, Hicks J, Miller FW, et al. Magnetic resonance imaging detection of occult skin and subcutaneous abnormalities in juvenile dermatomyositis. Arthritis Rheum. 2000;43:1866-73.
- 2. Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-72.
- 3. Jorizzo JL. Dermatomyositis. In: Bologna J, Jorizzo JL, Rapini RP, editors. Dermatology. London: Mosby; 2003. p.615-23.
- 4. Wagner E. Fall einer seltenen Muskelkrankheit. Deutsch Arch Heilk.1863;4:282.
- 5. Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920.
- 6. Garcia Vázquez E, Gutiérrez Guisado J, Blanco García A. Presentación de ocho casos de dermatomiositis: existe asociación de esta entidad con neoplasia? Rev Clin Esp. 1998;198:217-20.
- 7. Dourmishev LA. Dermatomyositis associated with malig nancy. 12 case reports. Adv Exp Med Biol. 1999;455:193-9.
- 8. Boham A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-7,403-7.
- 9. Drake LA, Dinehart SM, Farmer ER, Goltz RW, Graham GF, Hordinsky MK, et al. Guidelines of care for dermato myositis. American Academy of Dermatology. J Am Acad Dermatol. 1996;34(Pt 1):824-9.
- 10. Callen JP. Dermatomyositis: diagnosis, evaluation and management. Minerva Med. 2002;93:157-67.
- 11. Callen JP. When and how should the patient with der matomyositis or amyopathic dermatomyositis be assessed for possible cancer? Arch Dermatol. 2002;138:969-71.
- 12. Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am. 2002;28:723-41.
- 13. Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-30.
- 14. Adams-Gandhi LB, Boyd AS, King LE Jr. Diagnosis and management of dermatomyositis. Compr Ther. 1996;22:156-64.
- 15. Trüeb RM. Dermatomyositis. Dermatol Ther. 2001;14:70-80.
- 16. Jorizzo JL. Dermatomyositis: practical aspects. Arch Dermatol. 2002;138:114-6.
- 17. Callen JP. Collagen vascular diseases. J Am Acad Dermatol. 2004;51:427-39.
- 18. Pellissier JF, Civatte M, Fernandez C, Bartoli C, Chetaille B, Schleinitz N, et al. La dermatomyosite et la polymyosite. Rev Neurol (Paris). 2002;158:934-47.
- 19. Shamim EA, Rider LG, Miller FW. Update on the genetics of the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2000;12:482-91.
- 20. Medsger TA Jr, Oddis CV. Classification and diagnostic criteria for polymyositis and dermatomyositis. J Rheumatol. 1995;22:581-5.
- 21. Davies MG, Hickling P. Familial adult dermatomyositis. Br J Dermatol. 2001;144:415-6.
- 22. Cherin P, Chosidow O, Herson S. Polymyosites et der matomyosites. Actualités. Ann Dermatol Venereol. 1995;122:447-54.
- 23. Pachman LM, Litt DL, Rowley AH, Hayford JR, Caliendo J, Heller S, et al. Lack of enteroviral RNA or bacterial DNA in magnetic resonance imaging-directed muscle biopsies from twenty children with active untreated juvenile dermatomyositis. Arthritis Rheum. 1995;38:1513-8.
- 24. Okada S, Weatherhead E, Targoff IN, Wesley R, Miller FW, International Myositis Collaborative Study Group. Global surface ultraviolet radiation intensity may modu late the clinical and immunologic expression of autoim mune muscle disease. Arthritis Rheum. 2003;48:2285-93.
- 25. Callen JP. Dermatomyositis. Lancet. 2000;355:53-7.
- 26. Hengstman GJ, van Venrooij WJ, Vencovsky J, Moutsopoulos HM, van Engelen BG. The relative preva lence of dermatomyositis and polymyositis in Europe exhibits a latitudinal. Ann Rheum Dis. 2000;59:141-2.
- 27. Houpt KR, Sontheimer RD. Autoimmune connective tissue disease and connective tissue disease-like illnesses after silicone gel augmentation mammoplasty. J Am Acad Dermatol. 1994;31:626-42.
- 28. Hanke CW, Thomas JA, Lee WT, Jolivette DM, Rosenberg MJ. Risk assessment of polymyositis/dermatomyositis after treatment with injectable bovine collagen implants. J Am Acad Dermatol. 1996;34:450-4.
- 29. Targoff IN. Humoral immunity in polymyositis/ der matomyositis. J Invest Dermatol. 1993;100(Suppl1):S116-23.
- 30. Englund P, Nennesmo I, Klareskog L, Lundberg IE. Interleukin-1alpha expression in capillaries and major histocompatibility complex class I expression in type II muscle fibers from polymyositis and dermatomyositis patients: important pathogenic features independent of inflammatory cell clusters in muscle tissue. Arthritis Rheum. 2002;46:1044-55.
- 31. Nyberg P, Wikman AL, Nennesmo I, Lundberg I. Increased expression of interleukin 1alpha and MHC class I in muscle tissue of patients with chronic, inactive polymyositis and dermatomyositis. J Rheumatol. 2000;27:940-8.
- 32. Werth VP, Callen JP, Ang G, Sulllivan KE. Associations of tumor necrosis factor and HLA polymorphisms with adult dermatomyositis: implications for a unique pathogenesis. J Invest Dermatol. 2002;119:617-20.
- 33. Caro I. Dermatomyositis as a systemic disease. Med Clin North Am. 1989;73:1181-92.
- 34. Watanabe C, Okubo Y, Ohi T, Koga M, Abe H, Tawara K, et al. A case of dermatomyositis associated with mechanic's hand. J Dermatol. 2000;27:711-6.
- 35. Nousari HC, Ha VT, Laman SD, Provost TT, Tausk FA. "Centripetal flagellate erythema": a cutaneous manifestation associated with dermatomyositis. J Rheumatol. 1999;26:692-5.
- 36. Lupton JR, Figueroa P, Berberian BJ, Sulica VI. An unusual presentation of dermatomyositis: the type Wong variant revisited. J Am Acad Dermatol. 2000;43(Pt2):908-12.
- 37. del Pozo J, Almagro M, Martínez W, Yebra-Pimentel MT, García-Silva J, Peña-Penabad C, et al. Dermatomyositis and mucinosis. Int J Dermatol. 2001;40:120-4.
- 38. Park JH, Lee CW. Concurrent development of dermato myositis and morphoea profunda. Clin Exp Dermatol. 2002;27:324-7.
- 39. Aguiar FCA Jr, Gonzaga HFS, Almeida OP, Spolidório LC, Jorge MA. Dermatomiosite: revisão de literatura. J Bras Med. 2002;83:30-4.
- 40. Ihn H, Asano Y, Kubo M, Yamane k, Jinnin M, Yazawa N, et al. Clinical significance of serum surfactant protein D (SP-D) in patients with polymyositis/ dermatomyositis: correlation with interstitial lung disease. Rheumatology (Oxford). 2002;41:1268-72.
- 41. Marie I, Lecom F, Hachulla E, Antonietti M, François A, Levesque H, et al. An uncommon association: celiac disease and dermatomyositis in adults. Clin Exp Rheumatol. 2001;19:201-3.
- 42. Backhouse O, Griffiths B, Henderson T, Emery P. Ophthalmic manifestations of dermatomyositis. Ann Rheum Dis. 1998;57:447-9.
- 43. Juárez M, Misischia R, Alarcón GS. Infections in systemic connective tissue diseases: systemic lupus erythemato sus, scleroderma, and polymyositis/ dermatomyositis. Rheum Dis Clin North Am. 2003;29:163-84.
- 44. Huber AM, Hicks JE, Lachenbruch PA, Perez MD, Zemel LS, Rennebohm RM, et al. Validation of the childhood health assessment questionnaire in the juvenile idio pathic myopathies. J Rheumatol. 2001;28:1106-11.
- 45. Jäger C, Sirvent N, Rabasse N, Soler C, Sebag F, Boutte P, et al. Dermatomyosite juvénile dans la region niçoise. Etude rétrospective 1991-2001. Ann Dermatol Venereol. 2002;129:1120-4.
- 46. Pachman LM . Juvenile dermatomyositis: immuno genetics, pathophysiology, and disease expression. Rheum Dis Clin North Am. 2002;28:579-602, vii.
- 47. Ramanan AV, Sawhney S, Murray KJ. Central nervous sys tem complications in two cases of juvenile onset dermatomyositis. Rheumatology. 2001;40:1293-8.
- 48. Ghali FE, Stein LD, Fine J-D, Burkes J, McCauliffe DP. Gingival telangiectases: an underappreciated physical sign of dermatomyositis. Arch Dermatol. 1999;135:1370-4.
- 49. Russo RAG, Katsicas MM, Dávila M, Ciocca M, Zelazko M. Cholestasis in juvenile dermatomyositis. Report of three cases. Arthritis Rheum. 2001;44:1135-42.
- 50. Plamondon S, Dent PB. Juvenile amyopathic dermato myositis: results of a case finding descriptive survey. J Rheumatol. 2000;27:2031-4.
- 51. Stonecipher MR, Jorizzo JL, Monu J, Walker F, Sutej PG. Dermatomyositis with normal muscle enzyme concentrations. Arch Dermatol. 1994;130:1294-9.
- 52. Airio A, Pukkala E, Isomäki H. Elevated câncer incidence in patients with dermatomyositis: a population based study. J Rheumatol. 1995;22:1300-3.
- 53. Azulay RD, Azulay DR, Abulafia LA. Sinais malignos na pele versus síndromes paraneoplásicas cutâneas: revisão. An Bras Dermatol. 2000;75:621-30.
- 54. Leandro MJ, Isenberg DA. Rheumatic diseases and malignancy - is there an association? Scand J Rheumatol. 2001;30:185-8.
- 55. Nakanishi K, Cualing H, Husseinzadeh N. Dermatomyositis as a presenting symptom of ovarian cancer. Obstet Gynecol. 1999;94:836-8.
- 56. Yazici Y, Kagen LJ. The association of malignancy with myositis. Curr Opin Rheumatol. 2000;12:498-500.
- 57. Hagman JH, Bianchi L, Capione E, Vidolin AP, Chimenti S. Dermatomyositis associated with ovarian transitional cell carcinoma. J Am Dermatol. 2001;45:642-3.
- 58. Tanabe S, Mitomi H, Sada M, Yamazaki I, Akaboshi T, Okayasu I, et al. Parathyroid hormone-related protein production by adenocarcinoma in Barrett's esophagus patient with dermatomyositis. Dig Dis Sci. 2001;46:1584-7.
- 59. Joseph JV, Turner KJ, Bramwell SP. Dermatomyositis: a rare initial presentation of adenocarcinoma of the prostate. J Urol. 2002;168:637.
- 60. Lalla SC, Aldridge RD, Tidman MJ. Carcinoma of the penis presenting with dermatomyositis. Clin Exp Dermatol. 2001;26:556-8.
- 61. Mallon E, Osborne G, Dinneen M, Lane RJM, Glaser M, Bunker CB. Dermatomyositis in association with transi tional cell carcinoma of the bladder. Clin Dermatol. 1999;24:94-6.
- 62. Talanin NY, Bushore D, Rasberry R, Rudolph T, Tuli M, Friedman-Musicante R. Dermatomyositis with the fea tures of inclusion body myositis associated with carcino ma of the bladder. Br J Dermatol. 1999;141:926-30.
- 63. Federman DG, Radonich M, Kirsner RS. Fatal bladder cancer and dermatomyositis. South Med J. 2000;93:492-3.
- 64. Rankin WR, Herman JR. Rapidly progressive transition al cell carcinoma associated with dermatomyositis. J Urol. 2002;167(Pt 1):639-40.
- 65. Borgia F, Vaccaró M, Guarneri F, Cannavó SP, Guarneri B. Dermatomyositis associated with IgG myeloma. Br Jr Dermatol. 2001;144:186-221.
- 66. Hu WJ, Chen DL, Min HQ. Study of 45 cases of nasopha ryngeal carcinoma with dermatomyositis. Am J Clin Oncol. 1996;19:35-8.
- 67. Naschitz JE, Rosner I, Rozenbaum M, Zuckerman E, Yeshurun D. Rheumatic syndromes: clues to occult neo plasia. Semin Arthritis Rheum. 1999;29:43-55.
- 68. Gallais V, Crickx B, Belaich S. Facteur pronostiques et signes prédictifs de câncer au cours de la dermat omyosite de l'adulte. Ann Dermatol Venereol. 1996;123:722-6.
- 69. Basset-Seguin N, Roujeau JC, Gherardi R, Guillaume JC, Revuz J, Touraine R. Prognostic factors and predictive signs of malignancy in adult dermatomyositis. A study of 32 cases. Arch Dermatol. 1990;126:633-7.
- 70. Cox NH. Allergy to benzalkonium chloride simulating dermatomyositis. Contact Dermatitis. 1994;31:50-1.
- 71. Targoff IN. Update on myositis-specific and myositisassociated autoantibodies. Curr Opin Rheumatol. 2000;12:475-81.
- 72. Hausmanowa- Petrusewicz I, Kowalska- Oledzka E, Miller FW, Jarzabek- Chorzelska M, Targoff IN, Blaszczyk- Kostanecka M, et al. Clinical, serologic, and immuno genetic features in Polish patients with idiopathicinflammatory myopathies. Arthritis Rheum. 1997;40:1257-66.
- 73. Iannone F, Cauli A, Yanni G, Kingsley GH, Isenberg DA, Corrigall V, et al. T-lymphocyte immunophenotyping in polymyositis and dermatomyositis. Br J Rhematol. 1996;35:839-45.
- 74. Park JH, Olsen NJ, King L Jr, Vital T, Buse R, Kari S, et al. Use of magnetic resonance imaging and P-31 magnetic resonance spectroscopy to detect and quantify muscle dysfunction in the amyopathic and myopathic variants of dermatomyositis. Arthritis Rheum. 1995;38:68-77.
- 75. Hunger RE, Dürr C, Brand CU. Cutaneous leukocytoclastic vasculitis in dermatomyostis suggestts malignancy. Dermatology. 2001;202:123-6.
- 76. Oddis CY. Current approach to the treatement of polymyositis and dermatomyositis. Curr Opin Rheumatol. 2000;12:492-7.
- 77. Pelle MT, Callen PJ. Adverse cutaneous reactions to hydroxychloroquine are more common in patients with cutaneous lupus erythematosus. Arch Dermatol. 2002;138:1231-3; discussion 1233.
- 78. Cohen JB. Cutaneous involvement of dermatomyositis can respond to dapsone therapy. Int J Dermatol. 2002;41:182-4.
- 79. Gelber AC, Nousari HC, Wigley FM. Mycophenolate mofetil in the treatment of severe skin manifestations of dermatomyositis: a series of 4 cases. J Rheumatol. 2000;27:1542-5.
- 80. Hollar CB, Jorizzo JL. Topical tacrolimus 0.1% ointment for refractory skin disease in dermatomyositis: a pilot study. J Dermatolog Treat. 2004;15:35-9.
- 81. Chung YL, Alexanderson H, Pipitone N, Morrison C, Dastmalchi M, Stahl-Hallengren C, et al. Creatine sup plements in patients with idiopathic inflammatory myopathies who are clinically weak after conventional pharmacologic treatment: six-month, double-blind, ran domized, placebo-controlled trial. Arthritis Rheum. 2007;57:694-702.
- 82. Calvo Pulido M, Boixeda De Miquel P, Martín Saez E, Fernandez Guarino M, García-Millán C. Treatment of Gottron papules of dermatomyositis with pulsed dye laser. Eur J Dermatol. 2006;16:702-3.
- 83. Quain RD, Werth VP. Management of cutaneous der matomyositis: current therapeutic options. Am J Clin Dermatol. 2007;7:341-51.
- 84. Dold S, Justiniano ME, Márquez J, Espinoza LR. Treatment of early and refractory dermatomyositis with infliximab: a report of two cases. Clin Rheumatol. 2007;26:1186-8.
- 85. Chung L, Genovese MC, Fiorentino DF. A pilot trial of rituximab in the treatment of patients with dermato myositis. Arch Dermatol. 2007;143:763-7.
- 86. Dinh HV, McCormack C, Hall S, Prince HM. Rituximab for the treatment of the skin manifestations of dermatomyositis: a report of 3 cases. J Am Acad Dermatol. 2007;56:148-53.
- 87. Chen YJ, Wu CY, Shen JL. Predicting factors of malignan cy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol. 2001;825-31.
Datas de Publicação
-
Publicação nesta coleção
01 Ago 2008 -
Data do Fascículo
Jun 2008
Histórico
-
Recebido
29 Maio 2008 -
Aceito
29 Maio 2008