Resumos
A leucemia/linfoma de células T do adulto (ATL) é tipo agressivo de doença linfoproliferativa causada pelo vírus linfotrópico para células T humanas (HTLV-I), geralmente fatal e que não responde a quimioterapia. Classifica-se em formas aguda, crônica, linfomatosa e indolente (smoldering). Outra forma clínica, a tumoral primária de pele, com características diferentes, foi sugerida recentemente. As formas aguda, linfomatosa e tumoral primária de pele são as de pior prognóstico. Os critérios diagnósticos de ATL são: sorologia positiva para o HTLV-I; diagnóstico citológico ou histológico de leucemia/linfoma de células T, CD4+/CD25+; presença de linfócitos T anormais em sangue periférico; confirmação de integração monoclonal do DNA proviral do HTLV-I. Há lesões de pele em cerca de 70% dos casos,que podem ser primários (formas indolente e tumoral primária da pele) ou secundários. As lesões cutâneas são múltiplas, sendo as mais freqüentes a eritrodermia, as pápulas e as placas. A ATL não tem aspecto histológico característico, podendo apresentar padrões superponíveis ao linfoma periférico T não especificado, à micose fungóide ou ao linfoma anaplásico de grandes células. O padrão imuno-histoquímico pode também simular o de outros tipos de linfoma T. Por esse motivo, é muito importante que no Brasil seja solicitada sorologia para o HTLV-I em todos os casos de leucemia e/ou linfoma de células T maduras.
Leucemia; Linfoma anaplásico de grandes células; Linfoma cutâneo de células T; Micose fungóide; Paraparesia tropical espástica
Adult T cell leukemia/lymphoma (ATL) is an aggressive type of lymphoproliferative disease associated with the human T-cell lymphotropic virus type I (HTLV-I) that is characterized by a short survival time and absence of response to chemotherapy. ATL is classified into four clinical types: acute, chronic, lymphoma, and smoldering. Another clinical form of ATL, the primary cutaneous tumoral,with diverse characteristics, has been recently suggested. Patients with acute, lymphoma and primary cutaneous tumoral types have a poor prognosis. The diagnostic criteria of ATL consist of: positive serology for HTLV-I; cytologic or histologic confirmation of CD4+/CD25+ T-cell leukemia/lymphoma; abnormal T lymphocytes in peripheral blood; and confirmation of monoclonal integration of HTLV-I proviral DNA. There is skin involvement in around 70% of ATL cases, which could be primary (smoldering and primary cutaneous tumoral) or secondary. The skin lesions are multiple, erythroderma, papules and plaques being the most common. ATL has no characteristic histological pattern, and may present patterns that could superimpose nonspecific peripheral T-cell lymphoma, mycosis fungoides or anaplastic large cell lymphoma. The immunohistochemistry pattern may also be similar to that of other T-cell lymphomas. Thus, it is very important that in Brazil HTLV-I infection be investigated in all mature T-cell leukemias/lymphomas.
Adult T-cell leukemia; Anaplastic large-cell lymphomas; HTLV-I-associated myelopathy/tropical spastic paraparesis (HAM/TSP); Mycosis fungoides; T-cell cutaneous lymphoma
ARTIGO DE REVISÃO
Leucemia/linfoma de células T do adulto* * Trabalho realizado no Serviço de Patologia do Hospital Universitário Prof. Edgard Santos da Universidade Federal da Bahia (UFBA); Laboratório de Patologia Experimental (Lapex) do Centro de Pesquisas Gonçalo Moniz – Fundação Oswaldo Cruz (Fiocruz), Salvador (BA), Brasil. Conflito de interesse: Nenhum Suporte financeiro: Conselho Nacional de Pesquisa (CNPq); Fundação de Apoio à Pesquisa no Estado da Bahia (FAPESB).
Adult T-cell leukemia/lymphoma* * Trabalho realizado no Serviço de Patologia do Hospital Universitário Prof. Edgard Santos da Universidade Federal da Bahia (UFBA); Laboratório de Patologia Experimental (Lapex) do Centro de Pesquisas Gonçalo Moniz – Fundação Oswaldo Cruz (Fiocruz), Salvador (BA), Brasil. Conflito de interesse: Nenhum Suporte financeiro: Conselho Nacional de Pesquisa (CNPq); Fundação de Apoio à Pesquisa no Estado da Bahia (FAPESB).
Achiléa L. BittencourtI; Lourdes FarréII
IDoutora em medicina; pesquisadora sênior do Conselho Nacional de Pesquisa (CNPq); professora de patologia da Universidade Federal da Bahia (UFBA) – Salvador (BA), Brasil
IIDoutora em bioquímica; pesquisadora do Laboratório de Patologia Experimental (Lapex) do Centro de Pesquisas Gonçalo Moniz – Fundação Oswaldo Cruz (Fiocruz), Salvador (BA), Brasil; pesquisadora do Conselho Nacional de Pesquisa (CNPq)
Endereço para correspondência/ Mailing Address Endereço para correspondência/ Mailing Address: Achiléa L. Bittencourt Hospital Universitário Prof. Edgard Santos, Universidade Federal da Bahia (UFBA) Rua Dr Augusto Viana, s/n Canela 40110 060 - Salvador – BA E-mail: achilea@uol.com.br
RESUMO
A leucemia/linfoma de células T do adulto (ATL) é tipo agressivo de doença linfoproliferativa causada pelo vírus linfotrópico para células T humanas (HTLV-I), geralmente fatal e que não responde a quimioterapia. Classifica-se em formas aguda, crônica, linfomatosa e indolente (smoldering). Outra forma clínica, a tumoral primária de pele, com características diferentes, foi sugerida recentemente. As formas aguda, linfomatosa e tumoral primária de pele são as de pior prognóstico. Os critérios diagnósticos de ATL são: sorologia positiva para o HTLV-I; diagnóstico citológico ou histológico de leucemia/linfoma de células T, CD4+/CD25+; presença de linfócitos T anormais em sangue periférico; confirmação de integração monoclonal do DNA proviral do HTLV-I. Há lesões de pele em cerca de 70% dos casos,que podem ser primários (formas indolente e tumoral primária da pele) ou secundários. As lesões cutâneas são múltiplas, sendo as mais freqüentes a eritrodermia, as pápulas e as placas. A ATL não tem aspecto histológico característico, podendo apresentar padrões superponíveis ao linfoma periférico T não especificado, à micose fungóide ou ao linfoma anaplásico de grandes células. O padrão imuno-histoquímico pode também simular o de outros tipos de linfoma T. Por esse motivo, é muito importante que no Brasil seja solicitada sorologia para o HTLV-I em todos os casos de leucemia e/ou linfoma de células T maduras.
Palavras-chave: Leucemia/linfoma de células T do adulto; Linfoma anaplásico de grandes células; Linfoma cutâneo de células T; Micose fungóide; Paraparesia tropical espástica
ABSTRACT
Adult T cell leukemia/lymphoma (ATL) is an aggressive type of lymphoproliferative disease associated with the human T-cell lymphotropic virus type I (HTLV-I) that is characterized by a short survival time and absence of response to chemotherapy. ATL is classified into four clinical types: acute, chronic, lymphoma, and smoldering. Another clinical form of ATL, the primary cutaneous tumoral,with diverse characteristics, has been recently suggested. Patients with acute, lymphoma and primary cutaneous tumoral types have a poor prognosis. The diagnostic criteria of ATL consist of: positive serology for HTLV-I; cytologic or histologic confirmation of CD4+/CD25+ T-cell leukemia/lymphoma; abnormal T lymphocytes in peripheral blood; and confirmation of monoclonal integration of HTLV-I proviral DNA. There is skin involvement in around 70% of ATL cases, which could be primary (smoldering and primary cutaneous tumoral) or secondary. The skin lesions are multiple, erythroderma, papules and plaques being the most common. ATL has no characteristic histological pattern, and may present patterns that could superimpose nonspecific peripheral T-cell lymphoma, mycosis fungoides or anaplastic large cell lymphoma. The immunohistochemistry pattern may also be similar to that of other T-cell lymphomas. Thus, it is very important that in Brazil HTLV-I infection be investigated in all mature T-cell leukemias/lymphomas.
Keywords: Adult T-cell leukemia/lymphoma; Anaplastic large-cell lymphomas; HTLV-I-associated myelopathy/tropical spastic paraparesis (HAM/TSP); Mycosis fungoides; T-cell cutaneous lymphoma
INTRODUÇÃO
O vírus linfotrópico para células T humanas tipo I (Human T-cell lymphotropic vírus – HTLV-I) foi descoberto em 1980, isolado de células derivadas de paciente com linfoma cutâneo e, logo a seguir, relacionado à leucemia/linfoma de células T do adulto (adult T-cell leukemia/lymphoma – ATL), doença descrita três anos antes no Japão. Pouco depois da descoberta do HTLV-I, foi isolado outro retrovírus, o HTLV-II, que tem semelhança com o HTLV-I em 66% das seqüências genômicas, havendo reações sorológicas cruzadas entre eles.1
Antes de serem apresentados os diferentes aspectos da ATL, cabe fazer considerações sobre o HTLV-I, seus meios de transmissão, o diagnóstico da infecção e as demais doenças associadas a esse vírus.
O VÍRUS LINFOTRÓFICO DE CÉLULAS T HUMANAS TIPO I
O HTLV-I é endêmico, principalmente no sudoeste do Japão, nas ilhas do Caribe, na África Central, na América Central e no sudeste dos Estados Unidos. Na América do Sul a prevalência dessa infecção é um pouco menor. Estima-se que haja no mundo cerca de 15 a 20 milhões de portadores, mas as taxas de soroprevalência diferem em diferentes regiões. Estudo de soroprevalência do HTLV-I em doadores de sangue de 26 capitais brasileiras mostrou que as maiores freqüências foram observadas em São Luís (10.0/1.000), Salvador (9.4/1.000), Belém (9.1/1.000) e Recife (7.5/1.000). Em Salvador, estudo de soroprevalência mostrou, na população em geral, taxa de 1,8%, sendo mais elevada em mulheres (2%) do que em homens (1,2%).2
O HTLV-I é vírus envelopado que possui, no interior de seu capsídeo, duas cópias de RNA de fita simples, com polaridade positiva, associada a uma molécula de RNAt (RNA transportador), que serve como iniciador para a síntese de DNA. Além disso, contém ainda as enzimas transcriptase reversa, integrase e protease. O genoma do HTLV-I, com 9,03kb, possui os genes gag, pol e env, os quais codificam proteínas estruturais, flanqueados por duas seqüências denominadas seqüências terminais repetidas (long terminal repeats – LTRs). Tais seqüências contêm os promotores virais e outros elementos reguladores. O genoma do HTLV-I também contém a região pX que codifica as proteínas reguladoras tax, rex, HBZ, p12, p13, p30 e p21, as quais estão relacionadas com a oncogenicidade viral e proliferação das células infectadas.3
O HTLV-I pode infectar diversos tipos celulares como linfócitos B, linfócitos T, fibroblastos e monócitos, possuindo tropismo especial por células T CD4+. A transmissão do HTLV-I no organismo ocorre, principalmente, célula a célula, havendo transferência do material viral de célula infectada para célula não infectada. Nesse tipo de transferência, o contato entre a célula infectada e a não infectada leva a polarização do centro de organização do microtúbulo (microtubuleorganizing center – MTOC), formando uma "sinapse virológica" entre as células envolvidas. Dessa forma, proteínas e genomas virais acumulam-se na área de contato entre essas células e, posteriormente, ocorre a transferência do material viral para a célula não infectada.4
O aumento do número de células infectadas é a principal estratégia para a elevação da carga viral, de modo que a carga viral do HTLV-I é proporcional ao número de células infectadas.
Meios de transmissão
Transmite-se por via vertical, principalmente, pela amamentação, através de relação sexual com transmissão mais expressiva do homem para a mulher e parenteral, via transfusão sangüínea ou pelo uso compartilhado de instrumentos perfuro-cortantes contaminados.5 Meios de transmissão Transmite-se por via vertical, principalmente, pela amamentação, através de relação sexual com transmissão mais expressiva do homem para a mulher e parenteral, via transfusão sangüínea ou pelo uso compartilhado de instrumentos perfuro-cortantes contaminados.5
Diagnóstico da infecção pelo HTLV-I
O método diagnóstico de rotina mais usado para detecção de anticorpos anti-HTLV-I no soro é o imunoenzimático (enzyme-linked immunosorbent assay – Elisa) que deve ser confirmado pelo Western-blot, que permite a diferenciação entre os tipos I e II do HTLV. Em casos nos quais não é possível confirmar a infecção por essa técnica, deve ser usada a reação em cadeia da polimerase que detecta o DNA proviral.1
Doenças associadas ao HTLV-I
Muito embora a maioria dos portadores do HTLV-I não desenvolva doenças associadas, alguns deles podem manifestar enfermidades de difícil controle, como a leucemia/linfoma de células T do adulto (ATL), a paraparesia espástica tropical/mielopatia associada ao HTLV-I (HTLV-I – associated myelopathy/tropical spastic paraparesis – HAM/TSP) e a dermatite infecciosa associada ao HTLV-I (DIH). O HTLV-I tem ação muito lenta no organismo, e, por isso, as doenças que causa são, à exceção da DIH, de aparecimento muito tardio, mesmo quando a infecção ocorre nos primeiros anos de vida.1
A HAM/TSP é doença incapacitante, de evolução lenta e progressiva, envolvendo, sobretudo, a medula espinhal. Ocorre, na quarta e quinta décadas de vida, A dermatite infecciosa é um tipo de eczema infantil, descrito pela primeira vez na Jamaica e que ocorre em crianças infectadas verticalmente pelo HTLV-I. Já foram descritos casos de DIH que depois desenvolveram ATL.1
No Estado da Bahia têm-se observado estreita relação entre DIH, HAM/TSP e ATL. Já foram relatados vários casos de DIH em crianças e adolescentes, 30% dos quais evoluíram para a forma infanto-juvenil da HAM/TSP, e um deles também para ATL. 6-8 Por outro lado, de 52 casos de ATL com envolvimento da pele observados na Bahia, 37% tiveram história de doença compatível com de DIH na infância (Bittencourt, comunicação pessoal).
Em outras doenças causadas direta ou indiretamente pelo HTLV-I, incluem-se polimiosite, uveíte, artropatia, alveolite linfocitária, síndrome de Sjögren, tireoidite, doença de Behçet e polimiosite.1 Na infecção pelo HTLV-I, o sistema imunológico fica desregulado, com proliferação espontânea de linfócitos T e elevação simultânea das citocinas dos linfócitos T auxiliadores 1 (Th1) e dos linfócitos T auxiliadores 2 (Th2). Por esse motivo, os indivíduos infectados são mais susceptíveis a agentes infecciosos. Apresentam, com maior freqüência do que pessoas não infectadas, dermatomicoses, sarna norueguesa, estrongiloidíase, infecções bacterianas da pele e tuberculose.1
Têm sido referidas doenças neurológicas associadas ao HTLV-II, muito embora o papel desse vírus na patogênese dessas patologias ainda não esteja claro.9
LEUCEMIA/LINFOMA DE CÉLULAS T DO ADULTO (ATL)
A ATL constitui forma grave de leucemia/linfoma, que ocorre na vida adulta, não responde à quimioterapia e é, geralmente, fatal. Ocorre em cerca de 5% dos indivíduos infectados e manifesta-se, em geral, após longo período de latência. Esse período, no Japão, é de cerca de 60 anos, enquanto no Brasil a doença manifesta-se uma década mais cedo.10 No Estado da Bahia observou-se que um terço dos linfomas de células T maduras é associado a esse vírus.11 No Estado do Rio de Janeiro, essa freqüência é em torno de 26,5%.12 Acredita-se que a ATL ocorra quase exclusivamente em indivíduos infectados verticalmente.1
Ainda que pouco freqüente, a ATL tem sido observada muito mais precocemente, em crianças e adolescentes.1 Embora essa enfermidade seja muito agressiva, têm sido registrados casos com evolução muito prolongada.13,14
Aspecto curioso – e considerado raro na literatura 10 – observado no Estado da Bahia foi a associação elevada do ATL com HAM/TSP.
Os indivíduos soropositivos assintomáticos que apresentam integração monoclonal do DNA proviral nos linfócitos do sangue periférico são considerados casos pré-ATL. Essa condição ocorre em cerca de 1,7% dos portadores. Acompanhando 50 casos de pré-ATL durante 20 anos, Imaizumi et al.15 observaram que 21 (42%) evoluíram para ATL. Houve maior risco quando ocorreu associação com leucocitose.15
Aspectos clínicos
A diversidade das manifestações clínicas da ATL determinou a subdivisão da doença em quatro formas clínicas – aguda, crônica, linfomatosa e indolente (smoldering), todas com características específicas, descritas a seguir:
Forma indolente: observou-se presença de 5% ou mais de linfócitos T anormais no sangue periférico; ausência de linfocitose (< 4 x 109/litro) e de hipercalcemia. Desidrogenase lática (LDH) aumentada até 1,5 vez o valor normal; ausência de linfadenopatia, derrames cavitários, envolvimento de fígado, baço, sistema nervoso central, ossos e trato gastrointestinal. Pode haver comprometimento da pele e/ou dos pulmões. Nos pacientes com menos de 5% de linfócitos T anormais no sangue, é necessário que o estudo histológico de pele ou pulmão demonstre infiltração por células neoplásicas.16
Forma crônica: presença de linfocitose absoluta com mais de 4 x 109/litro; LDH até duas vezes o valor normal; ausência de hipercalcemia, ascite ou derrame pleural. Ausência de envolvimento do sistema nervoso central, ossos e trato gastrointestinal. Pode apresentar envolvimento linfonodal, de fígado, baço, pele e/ou pulmões. No sangue periférico, podem ser identificados 5% ou mais de linfócitos anormais.16
Forma linfomatosa: apresenta-se com linfadenomegalia, sem linfocitose e com 1% ou menos de linfócitos anormais no sangue periférico. É necessária a comprovação histológica de infiltração neoplásica nos linfonodos, associada ou não a envolvimento extranodal.16
Forma aguda: é a mais agressiva, cursando com linfadenomegalia, hepatosplenomegalia, lesões cutâneas, ósseas, gastrointestinais e do sistema nervoso central. Apresenta características que excluem os demais subtipos. Manifesta-se com leucemia aguda, numerosos linfócitos atípicos no sangue, hipercalcemia, lesões cutâneas e hepatosplenomegalia. Nessa forma clínica há evolução rápida para o óbito.16
Forma tumoral primária de pele: corresponde a forma clínica não incluída na classificação de Shimoyama et al.16 Apresenta tumores na pele e ausência de linfocitose, hipercalcemia, envolvimento linfonodal e de outros órgãos internos. Cursa com níveis de LDH pouco elevados.10 Nesta forma, os linfócitos anormais no sangue periférico mantêm-se abaixo de 5% (Bittencourt, dados pessoais).
Johno et al.17 descreveram a ATL primária da pele classificando-a em dois tipos, o tumoral e o eritematopapuloso, considerando que o tipo tumoral apresentava prognóstico pior. Como o tipo eritematopapuloso corresponde à forma indolente da classificação referida, com lesões restritas à pele, Bittencourt et al.10 sugeriram a inclusão na classificação de Shimoyama et al.16 apenas da forma tumoral primária da pele, a qual constitui tipo distinto de ATL, que deve ser diferenciado da forma indolente, pois apresenta curso bem mais agressivo.10
A classificação de Shimoyama et al.16 não é muito clara quanto à definição da forma indolente e, por esse motivo, tem sido interpretada na literatura de maneira diferente. Segundo esses autores, há dois padrões hematológicos da forma indolente, um com 5% ou mais de células anormais no sangue (não necessariamente células "em flor"), e outro com menos de 5% de células anormais, devendo ter também envolvimento maligno da pele e/ou dos pulmões. Muitos autores, no entanto, em suas revisões, consideram apenas este último padrão.18,19
Qualquer um dos tipos de ATL pode evoluir para a forma aguda, condição denominada "de crise".16 A ATL é muito letal, sendo que em 70 casos observados no Estado da Bahia, a mediana de tempo de sobrevida (MTS) foi de 12 meses, com apenas 13% dos pacientes ainda vivos na última avaliação. No entanto, as formas indolente e crônica apresentam sobrevida mais prolongada.
Envolvimento cutâneo
O envolvimento cutâneo pode ocorrer em todas as formas clínicas de ATL, com freqüência variando de 43 a 72%. No Estado da Bahia, encontrase envolvimento cutâneo em 67% dos casos, e todos os casos da forma indolente têm manifestações cutâneas e menos de 5% de células atípicas no sangue, o que indica serem primários de pele.10
Sabe-se que o critério para considerar um linfoma cutâneo primário é ser restrito à pele na época do diagnóstico. Assim sendo, a forma indolente com menos de 5% de linfócitos anormais no sangue e a forma tumoral primária de pele representam formas de ATL primárias cutâneas. É importante diferenciar as formas primárias das secundárias de pele, como se faz em relação aos demais linfomas.20 No entanto, na classificação da WHO/EORTC (World Health Organization/European Organization for Research and Treatment of Cancer), a ATL está referida como linfoma que só pode atingir a pele secundariamente.20
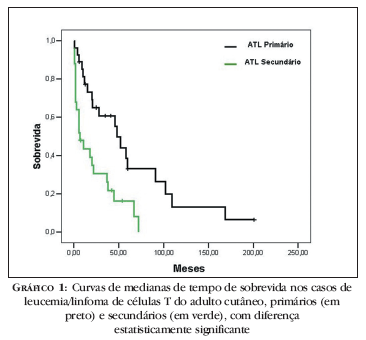
Em 52 casos de ATL com envolvimento cutâneo, comprovado por estudo histopatológico, observou-se que as formas primárias de pele tiveram MTS de 48 meses, enquanto as formas com envolvimento secundário tiveram apenas sete meses, e essa diferença foi estatisticamente significante (Gráfico 1). No entanto, na forma indolente observou-se MTS bem mais elevada (58 meses) do que na forma tumoral primária da pele (20 meses), o que confirma a importância de definir nitidamente esses dois tipos de ATL primários da pele (Bittencourt, dados pessoais). Os pacientes com doença primária da pele, formas indolente e tumoral falecem devido às infecções associadas, causas não relacionadas ao HTLV-I e, bem menos freqüentemente, evolução para a forma aguda.10
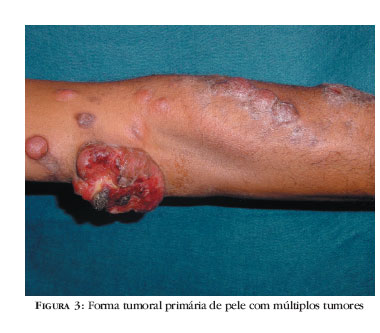
A ATL manifesta-se, na pele sob forma de eritrodermia (Figura 1), placas, pápulas (Figura 2), nódulos, tumores (Figura 3) e ou máculas. As lesões são sempre múltiplas e generalizadas em 50% dos casos. Dentre as mais freqüentes, destacam-se a eritrodermia, as placas e as pápulas. Não se observa correlação entre tipo clínico e morfologia das lesões, com exceção dos tumores que só são vistos nas formas clínicas mais agressivas (aguda, linfomatosa e tumoral primária da pele) e a eritrodermia que não é observada na forma tumoral primária da pele (Bitttencourt, dados pessoais).
Como já referido, o aspecto histopatológico da ATL é muito variado e pode ter aspectos superponíveis a outros linfomas T não associados ao HTLV-I. Contudo, de acordo com a classificação dos linfomas em geral da WHO21 e a classificação dos linfomas cutâneos da WHO/EORTC,20 todos os casos de leucemia/ linfomas associados ao HTLV-I, independentemente do padrão histopatológico, são classificados como ATL, não levando em consideração o fato de que esse diagnóstico só pode ser dado após estudos sorológicos, hematológicos ou moleculares porque a ATL não tem aspectos histopatológicos característicos. O patologista geral ou o dermatopatologista, sem conhecer o resultado desses estudos, diagnosticará a ATL como outro tipo de linfoma T.10 Por esse motivo, é recomendável que no Brasil a sorologia para o HTLVI seja solicitada em todos os casos de leucemia/linfoma de células T maduras.
Aspectos histológicos e imunofenotípicos
A ATL é reconhecida como entidade na classificação dos linfomas em geral da WHO, sendo considerada neoplasia de células T maduras, disseminada e leucêmica, e associada à infecção pelo HTLV-I.21 A ATL tem, mais freqüentemente, aspecto morfológico de linfoma T periférico não especificado, mas pode mostrar, microscopicamente, padrões superponíveis a outros tipos de linfoma T, sobretudo a micose fungóide e o linfoma anaplásico de células grandes.10
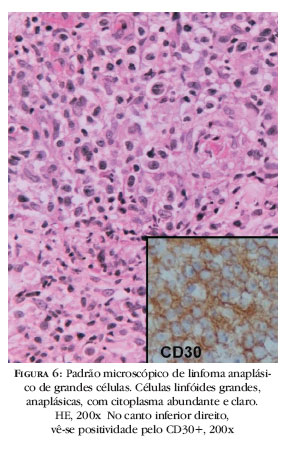
A principal característica morfológica dos linfomas T periféricos não especificados consiste na variação do tamanho das células e no acentuado pleomorfismo nuclear (Figura 4). Esse linfoma corresponde a diversos subtipos morfológicos de classificações antigas, incluindo o linfoma T pleomórfico.22 Nos casos com histologia de micose fungóide, os aspectos histológicos são superponíveis aos dos pacientes com igual tipo de linfoma e não infectados, com presença de células cerebriformes, epidermotropismo, obliteração da basal e abscessos de Pautrier (Figura 5). Como se vê na figura 4, nos linfomas T periféricos não especificados também se observam epidermotropismo de linfócitos e abscessos de Pautrier. Em estudo comparativo entre aspectos histopatológicos de linfoma T não especificado e de micose fungóide de indivíduos soropositivos e soronegativos para o HTLV-I, Bittencourt e cols.23 não encontraram diferenças significativas. No linfoma anaplásico de células grandes, as células neoplásicas são geralmente grandes e coesas, com abundante citoplasma e núcleos volumosos, arredondados e com nucléolo único e central (Figura 6).
O imunofenótipo da ATL é, geralmente, C D 3 + / C D 4 + / , C D 5 + / C D 7 - / C D 8 - / C D 2 0 - /CD25+/CD45RO+/CD79a-. No entanto, podem ocorrer casos CD4+ CD8+, CD4- CD8+ e CD4-CD8-. Nos casos com morfologia de linfoma anaplásico de células grandes as células malignas são CD30+.10 É importante determinar o índice proliferativo do linfoma, o que pode ser obtido por imuno-histoquímica com o marcador Ki-67. Esse dado é importante na avaliação prognóstica do paciente.10
Diagnóstico
Os critérios para o diagnóstico da ATL são: 1. presença de anticorpos séricos para o HTLV-I; 2. comprovação citológica ou histopatológica de leucemia e ou linfoma de células T maduras com antígenos de superfície CD4+/CD25+; 3. presença de linfócitos T anormais em sangue periférico, principalmente das células "em flor", que são consideradas características de ATL;24 4. demonstração da integração monoclonal do HTLV-I.24 O diagnóstico da ATL pode, muitas vezes, ser feito sem a demonstração da integração proviral; no entanto, em casos atípicos é importante essa demonstração. Lamentavelmente, na América Latina essa avaliação só é feita em raros laboratórios de pesquisa, não sendo ainda accessível a todos os profissionais envolvidos no diagnóstico dessa enfermidade.
A definição da forma clínica de ATL é de grande importância para orientar o tratamento e prever a evolução, porque há diferenças quanto ao prognóstico entre as diferentes formas clínicas.10
É importante chamar a atenção para dificuldades diagnósticas que podem ocorrer em relação à forma indolente, cujo diagnóstico baseia-se, freqüentemente, no encontro de um linfoma T em biópsia cutânea. Em algumas lesões com infiltração de linfócitos T, o quadro histológico pode não ser conclusivo de linfoma pela falta de atipias e, nessa circunstância, a imuno-histoquímica, em geral, não ajuda. Nesses casos, quando possível, deve-se realizar a análise do rearranjo dos genes que codificam o receptor para células T (TCR), a qual permite verificar se existe expansão monoclonal dos linfócitos T no sangue periférico ou nas lesões de pele, permitindo a diferenciação entre linfoma e algum processo inflamatório ou hiperplásico da pele. Convém ressaltar que a infecção pelo HTLV-I em portadores ou em pacientes com HAM/TSP pode causar lesões cutâneas eritematosas e ou descamativas, inflamatórias, não infecciosas e não linfomatosas, muitas vezes com histologia suspeita de malignidade 25,26 e que devem ser diferenciadas de linfomas T cutâneos. Por outro lado, os pacientes com essa infecção freqüentemente apresentam escabiose e ou mesmo sarna norueguesa, condições que podem causar hiperplasia linfóide reacional que, muitas vezes, simula linfoma T cutâneo.27 Pela análise do rearranjo dos genes que codificam o TCR pode-se definir o diagnóstico diferencial entre essas condições.
Em áreas em que o HTLV-I é endêmico, deve-se diferenciar a ATL dos linfomas e/ou leucemias não relacionados ao vírus e ocorrendo coincidentemente em paciente infectado. Nesses casos, quando possível, pode-se recorrer à pesquisa de integração proviral do HTLV-I.18 É importante esse diagnóstico diferencial porque o prognóstico e o tratamento da ATL é diferente dos de outros linfomas T periféricos.10 Na figura 7 observa-se padrão monoclonal de integração viral em um caso de forma indolente, iniciado na infância e com evolução superior a 14 anos, que foi atípico quanto à evolução e ao início da doença, requerendo a realização desse teste para comprovar a relação do linfoma com o vírus.
Fatores prognósticos
Os principais indicadores de pior prognóstico, determinados em estudo de 854 pacientes, por análise multivariada, foram: estado avançado da doença, elevados níveis de LDH, idade > 40 anos, mais de três áreas envolvidas e hipercalcemia. Também foram associados com pior prognóstico, entre outros fatores, trombocitopenia, eosinofilia, envolvimento da medula óssea e elevados níveis de interleucina-5.18 Esses indicadores, em sua maioria, estão presentes na forma aguda, que é a de pior prognóstico. De referência à forma crônica da doença, pacientes que têm níveis elevados de LDH e de uréia e baixos níveis de albumina têm pior prognóstico.18
Em estudo realizado com 70 casos de ATL, por análise univariada, os fatores relacionados a pior prognóstico foram: formas clínicas aguda, linfomatosa e tumoral primária de pele, índice proliferativo maior que 18%, presença de células grandes na histologia e ausência de lesões cutâneas.10 O envolvimento cutâneo predominou nas formas de melhor prognóstico, tendo estado presente em todos os casos da forma indolente e em 90% dos casos da forma crônica. Considerou-se que esses achados influenciaram a correlação positiva entre ausência de lesões cutâneas e pior prognóstico.10
Patogênese
O processo oncogênico do HTLV-I consiste em várias etapas e é multifatorial (Figura 8). No interior da célula infectada, o RNA do HTLV-I é transcrito em DNA de fita dupla pela enzima transcriptase reversa e, posteriormente, transportado para o núcleo, onde o DNA viral se insere no DNA genômico da célula hospedeira, interrompendo as seqüências humanas. O DNA viral inserido de modo randômico no DNA genômico humano é denominado DNA proviral. A integração proviral é necessária para a eficiente expressão dos genes virais e para a replicação do vírus.
Após a integração, há expressão das proteínas virais nas células infectadas, tais como a tax, que é considerada a principal indutora das etapas iniciais da oncogênese nessa infecção, pois estimula a proliferação e inibe a apoptose da célula infectada, regulando vias celulares-chave no controle desses processos, como a via de AKT, NF-kB e p53.28 Como a proteína tax localiza-se também na membrana celular, sua expressão na superfície das células infectadas as torna alvo das células T citotóxicas. No entanto, para poder escapar do sistema imune, a expressão de tax é inibida. Alguns trabalhos oriundos do Japão mostram que a expressão de tax é baixa ou inexistente na ATL. Recentemente, foi descrito o gene viral HTLV-1 bZIP factor (HBZ) cujo mRNA transcrito é responsável pela expansão dos linfócitos infectados, enquanto a proteína HBZ inibe a expressão de tax, permitindo que as células infectadas escapem do sistema imune.4
O contínuo estímulo da proliferação dos linfócitos infectados, durante um longo período de latência, provoca o aparecimento e o acúmulo de alterações genéticas e epigenéticas no genoma humano, levando à transformação neoplásica do linfócito e dando lugar ao surgimento de um clone celular com elevada capacidade de proliferação e sobrevida.4 A expansão desse clone neoplásico leva ao desenvolvimento da ATL. Assim, as células da ATL originam-se a partir de célula única e constituem uma população monoclonal (Figura 9) em que todas as células contêm o DNA proviral do HTLV-I, sempre integrado no mesmo local do genoma humano. Essa condição que caracteriza a ATL é considerada integração monoclonal do HTLV-I.29 A demonstração da integração monoclonal proporciona o diagnóstico diferencial da ATL com outros linfomas T não relacionados ao vírus e que podem ocorrer em portadores do HTLV-I.
Tratamento
Diversos protocolos já foram descritos para o tratamento de ATL com resultados variáveis. Dentre eles destacam-se os que empregaram quimioterapia, ziduvidina (AZT)/interferon-α (IFN-α) e transplante hematopoiético de células-tronco. A escolha do tratamento depende da forma de ATL e da avaliação dos fatores prognósticos.18 Têm sido obtidos melhores resultados com a associação AZT/IFN-α do que com a quimioterapia. Essa associação dá bons resultados nas formas aguda, crônica e indolente, mas não beneficia os pacientes com a forma linfomatosa.30
Tradicionalmente, os pacientes com as formas indolente e crônica de bom prognóstico não são submetidos a tratamento, a não ser quando há progressão do quadro.18 No entanto, já se observou que tratando essas formas com a associação AZT/IFN-α obtém-se sobrevida geral de 100% em seguimento médio de 10 anos, sendo esses resultados superiores aos da conduta expectante.30□
Aprovado pelo Conselho Editorial e aceito para publicação em 30.07.2008.
- 1. Bittencourt AL, Primo J, de Oliveira MF. Manifestations of the human T-cell lymphotropic vírus type I infection in childhood and adolescence. J Pediatr. 2006;82:411-20.
- 2. Catalan-Soares BC, Proietti FA. HTLV-1 e 2: aspectos epidemiológicos. In: Cadernos Hemominas - HTLV. vol. XIII. Belo Horizonte: Fundação Hemominas; 2006. p.69-85.
- 3. Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer. 2007;7:270-80.
- 4. Bangham CR. The immune control and cell-to-cell spread of human T-lymphotropic virus type 1. J Gen Virol. 2003;84:3177-89.
- 5. Bittencourt AL. Vertical transmission of HTLV-I/II: a review. Rev Inst Med Trop Sao Paulo. 1998;40:245-51.
- 6. Oliveira Mde F, Brites C, Ferraz N, Magalhaes P, Almeida F, Bittencourt AL. Infective dermatitis associated with the human T cell lymphotropic vírus type I in Salvador, Bahia, Brazil. Clin Infect Dis. 2005;40:90-6.
- 7. Primo JR, Brites C, Oliveira Mde F, Moreno-Carvalho O, Machado M, Bittencourt AL. Infective dermatitis and human T cell lymphotropic vírus type 1-associatedmyelopathy/ tropical spastic paraparesis in childhood and adolescence. Clin Infect Dis. 2005;41:535-41.
- 8. Farré L, de Oliveira MdeF, Primo J, Vandamme AM, Van Weyenbergh J, Bittencourt AL. Early sequential development of infective dermatitis, human T cell lymphotropic vírus type 1-associated myelopathy, and adult T cell leukemia/lymphoma. Clin Infect Dis. 2008;46:440-2.
- 9. Araújo A, Hall WW.Human T-lymphotropic vírus type II and neurological disease. Ann Neurol. 2004;56:10-9.
- 10. Bittencourt AL, Barbosa HS, Vieira MG, Alves CRB, Farré L. Adult T-cell leukemia/lymphoma (ATL) in Bahia, Brazil: analysis of prognostic factors in a group of 70 patients. Am J Clin Pathol. 2007;128:875-82.
- 11. Barbosa HS. Linfomas e leucemias associados à infecção pelo HTLV-I no Estado da Bahia [Tese]. Salvador, Bahia: Universidade Federal da Bahia, 1997. 124p.
- 12. Pombo de Oliveira MS, Matutes E, Schultz T, Carvalho SM, Noronha H, Reaves JD, et al. T-cell malignancies in Brazil. Clinicopathological and molecular studies of HTLV-I positive and negative cases. Int J Cancer. 1995;60:823-7.
- 13. Bittencourt AL, Barbosa HS, Pimenta A, Farre L. A case of adult-T-cell leukemia/ lymphoma (ATL) with a survival of more than 13 years. Acta Oncol. 2008;47:981-3.
- 14. Bittencourt AL, Barbosa HS, Requião C, Silva AC, Vandamme AM, Van Weyenbergh J, et al. Adult T-cell Leukemia/Lymphoma with a mixed CD4+ and CD8+ phenotype and a indolent course. J Clin Oncol. 2007;25:2480-2.
- 15. Imaizumi Y, Iwanaga M, Tsukasaki K, Hata T, Tomonaga M, Ikeda S. Natural course of HTLV-1 carriers with mon oclonal proliferation of T lymphocytes ("pre-ATL") in a 20-year follow-up study. Blood. 2005;105:903-4.
- 16. Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the lymphoma study group (1984-87). Br J Haematol. 1991;79:428-37.
- 17. Johno M, Ohishi M, Kojo Y, Yamamoto S, Ono T. Cutaneous manifestations of adult T-Cell leukemia/lymphoma. In: Takatsuki K, Hinuma Y, Yoshida M, editors. Advances in adult T-Cell Leukemia and HTLV-I Research. Gann monograph on Cancer Research nº 19. Japan, Tokyo: Scientific Society Press, 1992. p.33-42.
- 18. Ratner L. Human T cell lymphotropic vírus-associated leukemia/lymphoma. Curr Opin Oncol. 2005;17:469-73.
- 19. Matutes E. Adult T-cell leukaemia/lymphoma. J Clin Pathol. 2007;60:1373-7.
- 20. Burg G, Kempf W, Cozzio A, Feit J, Willemze R, S Jaffe E, et al. WHO/EORTC classification of cutaneous lymphomas 2005: histological and molecular aspects. J Cutan Pathol. 2005;32:647-74.
- 21. Jaffe ES, Ralfkiaer. Mature T-cell and NK-cell neoplasms: introduction. In: Jaffe ES, Harris N, Stein H, Vardiman JW, editors. World Health Organization Classification of Tumours: pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2001. p.189-94.
- 22. Ralfkiaer E, Müller-Hermelink HK, Jaffe ES. Peripheral Tcell lymphoma unspecified. In: Jaffe ES, Harris N, Stein H, Vardiman JW, editors. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2001. p.227-9.
- 23. Bittencourt AL, Barbosa HS, Brites C, Ferraz N, Freitas V, Pereira Filho CS, et al. Clinicopathological aspects of HTLV- I positive and negative cutaneous T-cell lymphoma: a comparative study. Eur J Dermatol. 1997;7:283-9.
- 24. Takatsuki K, Matsuoka M, Yamaguchi K. Adult T-cell leukemia in Japan. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;13 Suppl1:S15-9.
- 25. Rueda R, Blank A. HTLV-I associated cutaneous manifes tations. In: Zaninovic V, editores. HTLV, truths and ques tions. Cali: Feriva, 1996. p. 212-22.
- 26. Lenzi ME, Cuzzi-Maya T, Oliveira AL, Andrada-Serpa MJ, Araújo AQ. Dermatological findings of human T lymphotropic vírus type 1 (HTLV-I) associated myelopathy/tropical spastic paraparesis. Clin Infect Dis. 2003;36:507-13.
- 27. Ploysangam T, Breneman DL, Mutasim DF. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38:877-95.
- 28. Matsuoka M. Human T-cell leukemia virus type I (HTLVI) infection and the onset of adult T-cell leukemia (ATL). Retrovirology. 2005;2:27-40.
- 29. Kamihira S, Sugahara K, Tsuruda K, Minami S, Uemura A, Akamatsu N, et al. Proviral status of HTLV-1 integrated into the host genomic DNA of adult T-cell leukemia cells. Clin Lab Haematol. 2005;27:235-41.
- 30. Bazarbachi A, Panelatti G, Ramos JC, Tortevoye P, Otrock Z, Taylor G, et al. A worldwide meta-analysis on the use of zidovudine and interferon-alpha for the treatment of adult T-cell leukemia/lymphoma. Blood. 2007;110:610-1.
Datas de Publicação
-
Publicação nesta coleção
16 Set 2008 -
Data do Fascículo
Ago 2008
Histórico
-
Aceito
30 Jul 2008 -
Recebido
30 Jul 2008