Resumos
A doença de Fabry é enfermidade de armazenamento lisossômico rara, ligada ao cromossomo-X, causada pela deficiência parcial ou completa da enzima alfagalactosidase A. O defeito resulta no acúmulo de globotriaosilceramida no endotélio vascular e tecidos viscerais, sendo a pele, o coração, os rins e o sistema nervoso central os mais afetados. As autoras realizam revisão da literatura relacionada a essa afecção e ressaltam que o reconhecimento precoce dos angioqueratomas e da hipoidrose constitui sinal-chave no diagnóstico dessa doença grave. Destacam também a necessidade de esses doentes serem avaliados por equipe multidisciplinar.
Alfagalactosidase; Angioceratoma; Doença de Fabry
Fabry disease is an uncommon, X-linked lysosomal storage disorder, caused by partial or complete deficiency of the enzyme a-galactosidase A. The defect leads to accumulation of uncleaved globotriaosylceramide on the vascular endothelium and visceral tissues, being the skin, heart, kidneys and central nervous system the most affected organs. We performed review of the literature related to the disease and emphasized that early recognition of angiokeratomas and hypohidrosis are key diagnostic signs of this serious disease. We also addressed the need of multidisciplinary assessment of these patients.
Alpha-galactosidase; Angiokeratoma; Fabry disease
ARTIGO DE REVISÃO
Doença de Fabry* * Trabalho realizado na Dermatologia Pediátrica, Hospital Ramos Mejía Buenos Aires, Argentina. Conflito de interesse: Nenhum Suporte financeiro: Nenhum Como citar este artigo: Boggio P, Luna PC, Abad ME, Larralde M. Doença de Fabry. An Bras Dermatol. 2009;84(4):367-76.
Paula BoggioI; Paula Carolina LunaII; María Eugenia AbadIII; Margarita LarraldeIV
IDermatologista, assistente da Seção de Dermatologia Pediátrica, Departamento de Dermatologia, Hospital General de Agudos J.M. Ramos Mejía Buenos Aires, Argentina
IIDermatologista, assistente da Seção de Dermatologia Pediátrica, Departamento de Dermatologia, Hospital General de Agudos J.M. Ramos Mejía e do Departamento de Dermatologia, Hospital Churruca Buenos Aires, Argentina
IIIDermatologista, assistente da Seção de Dermatologia Pediátrica, Departamento de Dermatologia, Hospital General de Agudos J.M. Ramos Mejía e do Departamento de Dermatologia, Hospital Alemán Buenos Aires, Argentina
IVDermatologista, doutora em medicina pela Universidade de Buenos Aires (UBA) e professora adjunta de dermatologia da UBA. Chefe da Seção de Dermatologia Pediátrica, Departamento de Dermatologia, Hospital de Agudos J.M. Ramos Mejía Buenos Aires, Argentina. Chefe do Departamento de Dermatologia, Hospital Alemán Buenos Aires, Argentina
Endereço para correspondência Endereço para correspondência: Paula Boggio República de la India 3135, 3º andar, apart. "A", CEP: 1425 - Ciudad Autónoma de Buenos Aires (CABA), Buenos Aires, Argentina Tel/Fax: +54 11 48010405 E-mail: paulaboggio@fibertel.com.ar; paulaboggio@hotmail.com
RESUMO
A doença de Fabry é enfermidade de armazenamento lisossômico rara, ligada ao cromossomo-X, causada pela deficiência parcial ou completa da enzima alfagalactosidase A. O defeito resulta no acúmulo de globotriaosilceramida no endotélio vascular e tecidos viscerais, sendo a pele, o coração, os rins e o sistema nervoso central os mais afetados. As autoras realizam revisão da literatura relacionada a essa afecção e ressaltam que o reconhecimento precoce dos angioqueratomas e da hipoidrose constitui sinal-chave no diagnóstico dessa doença grave. Destacam também a necessidade de esses doentes serem avaliados por equipe multidisciplinar.
Palavras-chave: Alfagalactosidase; Angioceratoma; Doença de Fabry
HISTÓRICO
A doença de Anderson-Fabry, também chamada de doença de Fabry (DF) ou angiokeratoma corporis diffusum universale (OMIM #301500) foi relatada de maneira independente e quase simultânea pelos dermatologistas William Anderson na Inglaterra e Johannes Fabry na Alemanha, em 1898.1-3
DEFINIÇÃO
A DF é considerada doença de depósito lisossômico. Trata-se de erro inato do metabolismo dos glicoesfingolipídeos, produzido por mutações do gene que codifica a enzima lisossômica α-galactosidase A (α-GAL). A redução ou ausência da atividade dessa enzima leva ao acúmulo progressivo de glicoesfingolipídeos neutros com resíduos terminais α-galactosil (sobretudo sob a forma de globotriasilceramida ou GL-3) no plasma e nos lisossomos das células endoteliais de variados orgãos, principalmente pele, rins, coração, olhos e cérebro, com o resultante aparecimento da doença.4-7
GENÉTICA
A herança da DF é ligada ao cromossomo X. O gene afetado chama-se GALA e encontra-se na região Xq221 do cromossomo X. O GALA tem cerca de 12kb de comprimento e contém cerca de sete éxons que variam entre 92 e 291 pares de bases.8 O defeito genético que produz a doença é extremamente heterogêneo, e atualmente foram identificadas mais de 300 mutações.4,8 A maioria das famílias possui "mutações privadas", ou seja, mutações específicas para essa determinada família.4,9
Em doentes do sexo masculino, hemozigotos, o gene do Fabry tem alta penetrância, e a maioria apresenta o chamado fenótipo clássico da doença. Em doentes do sexo feminino, heterozigotas, a doença tem expressividade variável por causa da inativação aleatória de um dos cromossomos X (hipótese de Lyon). Atualmente as heterozigotas não são consideradas simples portadoras, já que a maioria apresenta alterações sistêmicas relacionadas à doença e aumento da morbidade e mortalidade. Portanto, a transmissão da DF de forma recessiva ligada ao X, deveria ser desconsiderada. 10-12
O grau de correlação existente entre o genótipo e o fenótipo da doença é motivo de controvérsia. Por exemplo, diferentes mutações estão associadas a fenótipos similares, enquanto membros de uma família que compartilha a mesma mutação apresentam diferentes fenótipos.7,13
Quando se suspeita do diagnóstico de DF, a pesquisa da história familiar é muito importante, já que a maioria dos casos ocorre de forma hereditária. Porém a ausência de dados familiares positivos não invalidam o diagnóstico, uma vez que mutaçoes têm sido documentadas.4,5,11
FISIOPATOGENIA
O defeito enzimático na DF leva ao acúmulo de glicoesfingolipídeos não clivados (particularmente GL-3) no plasma e nos lisossomos de inúmeros tipos celulares: células endoteliais, pericitos, células musculares lisas dos vasos sanguíneos, miócitos cardíacos, células epiteliais dos glomérulos e túbulos renais, células ganglionares do sistema nervoso autônomo, células da córnea, histiócitos e células do tecido conectivo. 5,14 Esse depósito de GL-3 nos lisossomos das células endoteliais, pericitos e células musculares lisas sanguíneas produz protuberâncias no lúmen dos vasos que causam estreitamentos e dilatações que progridem para isquemia e infarto,14 assim como também seu acúmulo em outros tipos de células leva a aumento do tamanho celular com aparecimento de organomegalias e disfunção visceral.15 Esse mecanismo fisiopatogênico explica a natureza multissistêmica dessa doença.
Em alguns tecidos específicos outros mecanismos adicionais contribuem para o aparecimento da doença. Acredita-se que, por exemplo, no coração, vias de sinalização determinantes de hipertrofia, apoptose, necrose e fibrose seriam ativadas e no sistema nervoso uma reação anormal da vasculatura e ativação da cascada da coagulação estariam envolvidas.16-18
EPIDEMIOLOGIA
A DF é mais frequente na raça branca, mas pode acometer todas as raças. Estima-se a incidência da doença em um caso a cada 40.000 homens ou um a cada 117. 000 nascidos vivos, representando, assim, a segunda alteração mais frequente por acúmulo lisossômico nos humanos.15,19 É possível que a incidência esteja subestimada devido à existência das variantes intermediárias da doença, como também aos casos diagnosticados post mortem e aqueles diagnosticados no contexto de um familiar afetado.5
APRESENTAÇÃO CLÍNICA
Os sintomas e sinais clínicos da DF são muito heterogêneos e sutis no começo, o que muitas vezes dificulta ou retarda seu diagnóstico.
Hemozigotos: geralmente apresentam a forma clássica da doença com perda total da função da enzima. O começo dos sintomas ocorre na infância ou na adolescência, com parestesias crônicas e episódios de dor acral e/ou abdominal (crises do Fabry), intolerância ao calor, diminuição ou ausência de sudorese, presença de angioqueratomas (AC) na pele e/ou mucosas e córnea verticilata. Entre a terceira e a quarta década de vida ocorre aumento desses sintomas e aparecem os relacionados ao comprometimento sistêmico progressivo (alterações cardíacas, renais e cerebrais). 4,5 Na ausência de história familiar da doença, o diagnóstico geralmente é feito tardiamente (idade média de 29 anos), quando já se desenvolveu dano visceral irreversível.4 Formas mais leves da doença, as quais se apresentam tardiamente com afecção primária dos sistemas renal ou cardiovascular, são conhecidas como variantes renal ou cardíaca, respectivamente, ou formas atípicas da DF e ocorrem em doentes com atividade enzimática detectável.20-22 Formas de gravidade intermediária entre o fenótipo clássico e as variantes renal ou cardíaca foram descritas e chamadas de formas intermediárias.
Heterozigotas: a apresentação clínica da doença nas mulheres é muito variável, e pode ir de estado assintomático a quadro tão grave quanto ao que ocorre em homens.19
Pacientes que apresentam o antígeno sanguíneo B apresentam manifestações clínicas relativamente mais graves, uma vez que a degradação desse antígeno também é dependente da enzima α-GAL.23
ACHADOS DERMATOLÓGICOS
O achado mais característico da DF são os angioqueratomas (AK) cutâneos disseminados ou AK corporis diffusum. Os AK são lesões vasculares que abrangem um ou mais vasos sanguíneos dilatados na derme superior, diretamente abaixo da epiderme, em geral acompanhados de reação epidérmica caracterizada por acantose e/ou hiperqueratose.24 Clinicamente, apresentam-se como inúmeras pápulas de cor variável do vermelho ao preto, com superfície discretamente queratósica, e tamanho variável de um até 10mm de diâmetro. Tendem a dispor-se em grupos e habitualmente têm distribuição simétrica e preferencial pela área de pele que vai do umbigo às coxas (disposição conhecida como em "roupa de banho") (Figura 1,2, e 3 ).4,6 A ocorrência de AK em áreas de trauma, como cintura, cotovelos, tornozelos, superfície de extensão dos braços e coluna vertebral, indica que esse fator mecânico atuaria como seu possível desencadeante (fenômeno de Koëbner) (Figura 4).4,5 Os AK surgem na adolescência nos hemozigotos, porém não está estabelecido claramente o momento do surgimento em mulheres, embora costume ocorrer em idades mais avançadas.19,25 A quase totalidade dos homens com o fenótipo clássico da DF terá AK, porém é frequente que estejam ausentes nas variantes cardíaca e renal da doença. Até 30% das heterozigotas desenvolverão essas manifestações cutâneas características, mas geralmente são lesões isoladas e em distribuições atípicas. 5,10 O número de AK e sua extensão aumenta conforme o passar do tempo24,26 e nem sempre têm relação direta com a morbidade sistêmica.4,5,27 São lesões persistentes, porém alguns AK podem sofrer trombose e desaparecer espontaneamente.28
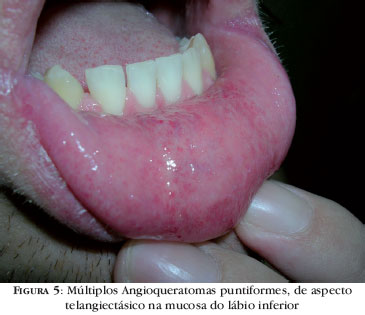
As mucosas também podem ser afetadas na DF. Com o avanço da doença é comum o aparecimento de AK nas mucosas, especialmente na mucosa oral, acometendo a porção anterior da mucosa jugal ou a mucosa labial e tendo aspecto telangiectásico (Figura 5).4,5
O sinal mais comum relatado pelos doentes é a diminuição da sudorese. Apresenta-se frequentemente na infância ou adolescência com pele seca e se traduz em intolerância ao calor e ao exercício, e às vezes febre sem causa aparente. 4,5 Propõe-se que a hipoidrose ou anidrose seja secundária ao dano seletivo de nervos autônomos, ao depósito de GL-3 nos ácinos sudoríparos e/ou à isquemia devida ao comprometimento dos vasos sanguíneos que nutrem tanto os nervos autônomos quanto as glândulas sudoríparas.29-31 Existem relatos de doentes com DF que apresentam hiperidrose, especialmente palmoplantar, que parece ser mais comum do que previamente acreditado, e mais prevalente nas mulheres.16,32-34
O pelo corporal pode ser acometido na DF na forma de hipotricose corporal difusa, pelo depósito direto de GL-3 nos folículos pilosos como também por alteração vascular de sua irrigação.33
Outra alteração comumente detectável ao exame físico dermatológico é a presença de edema nas pálpebras e extremidades.15 O linfedema das pernas é comum e se produz pelo depósito de GL-3 nos vasos linfáticos. 35 Raramente pode ser responsável pelo aparecimento de úlceras de estase recorrentes, que podem constituir manifestação inicial da doença.36
Existe um relato isolado de doente hemizigoto com DF que apresentou nódulos hipodérmicos doloridos que foram diagnosticados como poliarterite nodosa cutânea.37
ACHADOS NÃO DERMATOLÓGICOS
Neurológicos: O sintoma cardinal da DF é a parestesia crônica, que ocorre principalmente nos hemizigotos, acomete mãos e pés, começa na infância precoce e persiste até a vida adulta. Trata-se de uma dor ardente acompanhada de formigamento, que pode ser intermitente ou contínua, e que pode irradiar para áreas vizinhas.4,5 As acroparestesias compõem a principal causa de morbidade da doença durante as duas primeiras décadas da vida e, em alguns casos, levam à depressão e até mesmo a tentativas de suicídio. 29,38 Essa dor pode ser interrompida pelas chamadas crises de Fabry, que são episódios de dor aguda que duram desde minutos até dias, e que podem acompanhar- se de fadiga, febre de baixo grau e artralgias,4,5 sendo desencadeadas por estresse, fadiga, doença, aumento da temperatura externa e/ou exercício físico. 4,5 As acroparestesias e as crises tendem a sumir com o passar do tempo, possivelmente pela destruição completa dos filetes nervosos. 39,40 Na idade adulta a ocorrência de acidente vascular cerebral (AVC) se produz na idade média de 34 anos em hemizigotos e de 40 em heterozigotas, e se deve fundamentalmente à oclusão da microvasculatura ou à embolia.5 A doença cerebrovascular indica mau prognóstico; ela e a afecção cardiovascular são atualmente as maiores causas de morte desses doentes.41 Outras alterações neurológicas incluem anomalias auditivas (hipoacusia), sensoriais e vestibulares (vertigem e zumbido). 39
Oftalmológicos: Apresentam-se alterações conjuntivais que consistem em dilatações e tortuosidades vasculares, particularmente evidentes na conjuntiva bulbar inferior (Figura 6).4,15 As alterações retinianas são similares às anteriores e podem ser exacerbadas pela presença de doença renal e hipertensão arterial, constituindo achado muito comum nos hemizigotos. 29,42,43 As lesões do cristalino são menos frequentes e consistem em deposição de material granular e catarata subcapsular anterior.44 O achado ocular mais comum da DF é a córnea verticilata (opacidades amareladas caracterizadas por uma ou mais linhas irradiando de um ponto próximo ao centro da córnea), estando presente em quase todos os hemizigotos e em percentual que varia de 70 a 90% das heterozigotas. Essa alteração não afeta a visão. Como esse achado ocular é muito frequente, a oftalmoscopia de lâmpada de fenda é importante ferramenta no auxílio diagnóstico das mulheres com DF quando não se tem acesso aos estudos moleculares.44,45 Pode também ocorrer xeroftalmia de grau variável por diminuição da produção de lágrimas.15
Renais: A presença de poliúria por defeitos na concentração pode ser manifestação precoce do acometimento renal, mas é comumente ignorada. A maioria dos doentes com as formas clássicas desenvolve proteinúria na adolescência tardia, e é nesse momento que o dano renal é reconhecido.4,5 Ele é progressivo, evolui habitualmente para insuficiência renal crônica (IRC), que se apresenta entre a terceira e a quinta década de vida e é tratada com diálise crônica ou então com transplante renal.7,46 Após o transplante, a atividade da enzima do enxerto consegue metabolizar o GL-3 evitando o acometimento do rim transplantado. Logicamente, as heterozigotas não devem ser candidatas à doação.5 Anteriormente, quando a diálise e o transplante renal não eram realizados, a IRC era a principal causa de morte na DF e a expectativa de vida era de 41 anos aproximadamente.16 Na chamada variante renal da DF os doentes desenvolvem IRC em idades similares aos que apresentam o fenótipo clássico, mas na ausência de outras manifestações da doença.20
Cardiológicos: O comprometimento cardíaco é constante na DF. Produz-se pelo depósito de GL-3 no miocárdio, nas válvulas e no sistema de condução, o que explica a diversidade de sintomas, cuja expressão depende do sexo e da idade.5,47 As manifestações mais comuns são: hipertrofia do ventrículo esquerdo (HVE), insuficiência mitral, arritmias e doença coronariana arterial.14,47 A coexistência de HVE e alterações valvulares associa-se a doença mais grave e constitui fator prognóstico de gravidade da doença em geral.48
Alguns indivíduos com baixos níveis residuais da enzima α-GAL desenvolvem uma variante da doença que só tem expressão cardíaca, a chamada variante cardíaca do Fabry. Estima-se que entre três e 6% dos homens com cardiomiopatia hipertrófica não obstrutiva, considerada idiopática, tenham essa variante da DF.49
Outra manifestação menos conhecida dessa doença é alguma alteração do perfil lipídico que consiste em: colesterol total discretamente elevado, elevação do colesterol HDL e níveis normais de colesterol LDL e triglicérides.50
Heltianu et al. verificaram correlação entre a DF e anormalidades do gene eNOS. Esse gene determina a síntese de óxido nítrico, molécula essencial para as funções fisiológicas do sistema vascular, o qual é exclusivamente produzido pelas células endoteliais. Mutações do eNOS podem aumentar a predisposição ou intensificar doenças cardiovasculares, como, por exemplo, a DF. Constituem então um mecanismo adicional que explica por que um mesmo alelo para DF causa fenótipos diferentes entre os indivíduos portadores. 51
Gastrointestinais: Episódios de diarréia, náuseas, vômitos, dor pós-prandial e má absorção são as queixas mais comuns, e particularmente debilitantes nas crianças. Outros achados são: falência pancreática, divertículos jejunais e acalásia. Essas alterações se deveriam ao acúmulo de GL-3 nos vasos sanguíneos intestinais e nos gânglios autônomos.52
Fácies: Alguns doentes apresentam dismorfia facial de diferentes graus. Os achados são: lóbulos das orelhas proeminentes, sobrancelhas espessas, fronte deprimida, ângulo nasal pronunciado, nariz grande, ponte supraorbitária proeminente, base nasal larga e rasgos toscos.53,54
Estomatológicos: Achados na cavidade oral, além dos AK, incluem: xerostomia (por diminuição da secreção salivar), eritema e edema das papilas fungiformes, glossite, língua em empedrado, queilite granulomatosa e aumento na prevalência de cistos e pseudocistos dos seios maxilares.54
Psiquiátricos: Depressão, ideação suicida e demência. 5,15,55
Outros: Presença de AK na mucosa do trato respiratório e digestivo,15 maior incidência de hipotireoidismo subclínico, obstrução das vias aéreas, osteopenia até osteoporose, anemia, entre outros.5
DIAGNÓSTICO
A suspeita clínica da DF deverá ser confirmada da seguinte maneira:
Hemizigotos: o diagnóstico baseia-se na determinação dos níveis da α-GAL em lágrimas, plasma, leucócitos, cultura de fibroblastos cutâneos, ou, como foi recentemente descrito, em gotas de sangue seco colhidas em papel-filtro.56 Também pode ser feita a determinação da presença de GL-3 no sedimento urinário, plasma ou cultura de fibroblastos. 9
Heterozigotas: em alguns casos o diagnóstico pode ser feito como em hemizigotos. Mas, geralmente, esse teste é inadequado para triagem de mulheres, uma vez que elas apresentam inativação aleatória do cromossomo X entre suas células, havendo, assim, produção de quantidades suficientes da enzima no plasma ou tecidos. Portanto, o diagnóstico definitivo nas heterozigotas exige a identificação da mutação familiar específica no gene da GAL a partir de estudos de biologia molecular. 57,58
Diversos trabalhos demonstram que o diagnóstico definitivo da DF é feito, em média, 15 anos após o início dos sintomas.5
Como o acúmulo de GL-3 começa precocemente na vida intrauterina, é possível o diagnóstico prénatal através da demonstração da baixa atividade da α- GAL em biópsia de vilo coriônico ou cultura de células do líquido amniótico, quando se tratar de fetos com cariótipo XY.59 No caso de a mutação familiar da α-GAL ser conhecida, o estudo molecular pode suplantar ou confirmar o diagnóstico enzimático.
HISTOPATOLOGIA
Nas biópsias de diferentes tecidos dos doentes com DF, os achados histopatológicos consistem na presença de vacúolos citoplasmáticos contendo os lipídeos acumulados (preservação prévia dos tecidos com congelamento ou fixação em formol de cálcio 1%) na microscopia de luz.5 Na microscopia eletrônica se evidenciará a presença de inclusões lisossômicas, com a configuração lamelar concêntrica típica (alternância de bandas claras e escuras a cada 4-6nm), chamados de corpos de inclusão zebra-simil.5,60 Quando esses achados não são conclusivos, pode ser realizada imunoeletromicroscopia com anticorpos antiGL-3.5, 61
DIAGNÓSTICOS DIFERENCIAIS DERMATOLÓGICOS
O diagnóstico diferencial dos AK isolados se faz com: verruga vulgar, angioma senil ou rubi, granuloma piogênico, nevo de Spitz, queratose seborreica pigmentada e até melanoma.24 Também devem ser diferenciados de outros tipos de AK (Quadro 1).62
Os AK disseminados são fortemente sugestivos de DF, mas não são patognomônicos. Por esse motivo, deve-se afastar um amplo leque de doenças de depósito lisossômico, além de uma forma de AK corporis diffusum sem associação com nenhuma alteração enzimática conhecida, chamada de variante idiopática ou cutânea de AK corporis diffusum (Quadro 2).24,26,63
ACONSELHAMENTO GENÉTICO
Uma vez confirmado o diagnóstico de DF, o doente e seus familiares devem receber aconselhamento genético. Todas as filhas de um hemizigoto herdarão a doença, uma vez que herdarão o X que tem a mutação de seu pai, e nenhum dos seus filhos herdará a doença, já que eles vão herdar o cromossomo Y de seu pai. Metade dos filhos e metade das filhas de uma heterozigota serão acometidas, uma vez que ela tem um X com a mutação e outro sem.5,64
TRATAMENTO
O tratamento dessa complexa doença requer equipe multidisciplinar constituída por clínicos, dermatologistas, neurologistas, cardiologistas, nefrologistas e geneticistas experientes no assunto.
a. Terapia não específica
É tratamento de suporte, dirigido apenas ao controle dos sintomas e sinais presentes, e geralmente complementa a terapia específica.
Angioqueratomas: podem ser destruídos por diferentes métodos: eletrocoagulação, crioterapia, exérese cirúrgica ou Laser (Neodymium-YAG, pulsed dye, e outros). A laserterapia é o tratamento de escolha.33
Acroparestesias, crises de Fabry: os doentes são orientados para identificar e evitar os fatores precipitantes dos sintomas. Melhora parcial da dor é obtida com difenilidantoína, carbamazepina, gabapentina, oxacarbazepina ou topiramato.5,16,65
Doença vascular cerebral e retiniana: realiza- se prevenção com agentes antiplaquetários ou anticoagulantes. A proteção vascular pode ser intensificada com inibidores da enzima de conversão da angiotensina, estatinas e ácido fólico.5,65
Doença renal: controle da hipertensão arterial, diálise, até transplante renal.5 É indicado tratamento agressivo com inibidores da enzima conversora de angiotensina ou com bloqueadores dos receptores da angiotensina para reduzir a proteinúria.16,66
Doença cardíaca: controle das arritmias com drogas antiarrítmicas, marcapasso (quando houver indicação), até transplante cardíaco. Pacientes com doença coronariana podem ser candidatos a revascularização coronária.67
b. Terapia específica
Terapia de reposição enzimática (TRE)
Essa estratégia de tratamento tem como fundamento a descoberta de que as células podem incorporar uma enzima do meio extracelular e utilizá- la para seu metabolismo normal. A TRE para DF foi aprovada na Europa em 2001 e nos Estados Unidos em 2003. Atualmente existem duas α-GAL humanas disponíveis no comércio: a algasidase alfa (Replagal®, Transkaryotic Therapies Inc., Cambridge, Massachusetts), produzida por cultura de fibroblastos humanos acrescidos de promotores ativos para a transcrição do gene da α-GAL, aprovada na Europa; e a algasidase beta (Fabrazyme®, Genzyme Corp., Cambridge, Massachusetts), obtida por terapia recombinante de ovários de hamsters, aprovada na Europa e nos EUA.5,68 Ambas as proteínas são estrutural e funcionalmente semelhantes, têm atividade específica comparável e são administradas por via intravenosa a cada 15 dias. A dose é variável segundo o preparado: 0,2mg/kg/dose da algasidase alfa e 1mg/kg/dose da algasidase beta. A TRE é tratamento a ser utilizado por toda a vida, uma vez que a quantidade da enzima no plasma é rapidamente depletada, necessitando, assim, de infusões constantes.68 A tolerância à TRE é geralmente boa, com exceção de reações leves ou moderadas associadas à infusão, produto da formação de anticorpos IgG não neutralizantes.
É claro que a TRE reverte as anomalias metabólicas e várias das alterações patológicas da DF. A finalidade de sua implementação é prevenir o desenvolvimento de doença nos pacientes jovens e evitar, quando não reverter, a progressão da disfunção orgânica nos doentes mais velhos.68 Portanto, as recomendações atuais para o início da TRE são:16,69
Homizigotos:
- menores de 16 anos: assim que houver sintomas ou sinais.
- maiores de 16 anos: no momento do diagnóstico.
Heterozigotas: quando houver sintomas significativos ou acometimento de órgão nobre.
Estudos clínicos com ambas as preparações enzimáticas demonstraram diminuição da frequência das crises de dor, da massa cardíaca, e do depósito de GL-3 nos rins (com estabilização ou melhora da função renal nos casos de acometimento leve) e na pele. Existem evidências de que a TRE melhora a sudorese, os sintomas gastrointestinais, pulmonares e a audição. 5,16 No entanto, ainda não esta estabelecido claramente qual será o impacto a longo prazo da TRE na mortalidade da DF.
Terapia gênica: Essa técnica visa acrescentar um gene normal da α-GAL ao DNA do doente, passando este a produzir a enzima normalmente. Os métodos utilizados para introduzir genes estranhos nas células são classificados basicamente em sistemas virais (usam vetores virais como oncoretrovírus, lentivírus ou adenovírus) e não virais (liposomas contendo DNA). Ambos os sistemas de entrega têm sido testados no Fabry. A terapia gênica propõe o tratamento definitivo dessa doença, mas ainda está em fase experimental. 70
Outras: Existem outras modalidades específicas de tratamento da DF em estudo:
Chaperonas: é uma nova estratégia de realce enzimático, útil para os doentes que possuem variantes instáveis da α-GAL mutante, que, por defeitos de qualidade é retida no retículo endoplasmático, mas conservam atividade enzimática residual. Utilizam-se pequenas moléculas sintéticas que, atuando como chaperonas, resgatam a α-GAL residual transportandoa para os lissosomos. Essa terapia é administrada por via oral e ofereceria excelente complementação à TRE.71,72
Inibidores competitivos reversíveis da α-GAL: trata-se do uso de potentes inibidores competitivos da α-GAL, que, uma vez no interior das células, determinam aumento da atividade da enzima. Ao mesmo tempo, essas substâncias parecem acelerar o transporte, a maturação e a estabilidade da enzima mutante.73,74 Comportar-se-iam como chaperonas farmacológicas, sendo úteis apenas nos doentes com atividade enzimática residual.
Privação de substrato: baseia-se na inibição de etapas precoces na síntese de glicoesfingolipídeos com a consequente diminuição da formação e acúmulo de Gb3.75
CONCLUSÃO
A DF é quadro multissistêmico grave. A disponibilidade atual de tratamento específico obriga ao diagnóstico o mais precoce possível. No âmbito dermatológico, é preciso reconhecer os AK e a hipo-hidrose como dois sinais que auxiliam no diagnóstico.
AGRADECIMENTOS
À doutora Alice Lobo, do Departamento de Dermatologia do Hospital das Clínicas, da Faculdade de Medicina da Universidade de São Paulo São Paulo (SP), Brasil, por sua valiosa ajuda na correção do português.
Aprovado pelo Conselho Editorial e aceito para publicação em 27.12.2008.
- 1. Anderson W. A case of angeio-keratoma. Br J Dermatol. 1898;10:113-7
- 2. Fabry J. Ein beitrag zur kenntnis der purpura haemorrhagica nodularis (purpura papulosa haemorrhagica Hebrae). Arch Dermatol Syphilol (Berlin). 1898;43:187-200
- 3. Werninghaus K, Raab R, Palko M, Bhawan J. Punctate and linear angiectases: Anderson-Fabry disease (angiokeratoma corporis diffusum). Arch Dermatol. 1995;131:85-6
- 4. Larralde M, Boggio P, Amartino H, Chamoles N. Fabry disease: a study of 6 hemizygous men and 5 heterozygous women with emphasis on dermatologic manifestations. Arch Dermatol. 2004;140:1440-6
- 5. Larralde M, Luna P. Fabry disease. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller A, Leffell DJ, editors. Fitzpatrick's Dermatology in General Medicine. New York: McGraw-Hill; 2007. p.1281-8
- 6. Larralde de Luna M, García Díaz R, Sanchez G, Ilari R, Pierini AM, Campoy C, et al. Angioqueratoma "corporis diffusum"(enfermedad de Fabry) actualización: a propósito de 2 casos. Med Cutan Ibero-Latinoam. 1985;13:129-40
- 7. Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, et al. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002;81:122-38
- 8. Krawczak M, Ball EV, Fenton I, Stenson PD, Abeysinghe S, Thomas N, Cooper DN. Human gene mutation database-a biomedical information and research resource. Hum Mutat. 2000;15:45-51
- 9. Ashton-Prolla P, Ashley GA, Giugliani R, Pires RF, Desnick RJ, Eng CM. Fabry disease: comparison of enzymatic, linkage, and mutation analysis for carrier detection in a family with a novel mutation (30delG). Am J Med Genet. 1999;84:420-4
- 10. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769-75
- 11. Hasholt L, Sorensen SA, Wandall A, Andersen EB, Arlien-Soborg P. A Fabry's disease heterozygote with a new mutation: biochemical, ultrastructural, and clinical investigations. J Med Genet. 1990;27:303-6
- 12. Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. 2007;9:34-45
- 13. Matsuzawa F, Aikawa S, Doi H, Okumiya T, Sakuraba H. Fabry disease: correlation between structural changes in alpha-galactosidase, and clinical and biochemical phenotypes. Hum Genet. 2005;117:317-28
- 14. Desnick RJ, Joannou Y, Eng CM. κ-Galactosidase-Adeficiency: Fabry disease: nature of the accumulated glycosphingolipids. In: Scriver CH, Beaudet A, Valle D, editors. The metabolic and molecular basis of inherited disease. New York, NY: McGraw-Hill; 2001. p.3742-3
- 15. Möhrenschlager M, Henkel V, Ring J. Fabry disease: more than angiokeratomas. Arch Dermatol. 2004;140:1526-8
- 16. Zarate YA, Hopkin RJ. Fabry's disease. Lancet. 2008;372:1427-35
- 17. Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. 2007;93:528-35
- 18. Mehta A, Ginsberg L. Natural history of the cerebrovascular complications of Fabry disease. Acta Paediatr Suppl. 2005;94:24-7
- 19. Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J, et al. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001;24:715-24
- 20. Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a "renal variant"phenotype. Kidney Int. 2003;64:801-7
- 21. Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M, et al. An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288-93
- 22. von Scheidt W, Eng CM, Fitzmaurice TF, Erdmann E, Hübner G, Olsen EG, et al. An atypical variant of Fabry's disease with manifestations confined to the myocardium. N Engl J Med. 1991;324:395-9
- 23. Blom D, Speijer D, Linthorst GE, Donker-Koopman WG, Strijland A, Aerts JM. Recombinant enzyme therapy for Fabry disease: absence of editing of human alpha-galactosidase A mRNA. Am J Hum Genet. 2003;72:23-31
- 24. Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-82
- 25. Galanos J, Nicholls K, Grigg L, Kiers L, Crawford A, Becker G. Clinical features of Fabry's disease in Australian patients. Intern Med J. 2002;32:575-84
- 26. Caputo R, Ackerman BA, Sison-Torre EQ. Fabry's disease (Angiokeratoma corporis diffusum). In: Caputo R, Ackerman BA, editors. Pediatric Dermatology and Dermatopathology. Philadelphia: Lea & Febiger; 1994. p.285-93
- 27. Ries M, Schiffmann R. Fabry disease: angiokeratoma, biomarker, and the effect of enzyme replacement therapy on kidney function. Arch Dermatol. 2005;141:904-5
- 28. Bang DS, Choi YS, Song MS. Transepidermal elimination of thrombi in three cases of thrombotic angiokeratoma: an incidental histopathologic finding of angiokeratoma. J Dermatol. 1991;18:605-9
- 29. Kolodny EH, Pastores GM. Anderson-Fabry disease: extrarenal, neurologic manifestations. J Am Soc Nephrol. 2002;13:S150-3
- 30. Onishi A, Dyck PJ. Loss of small peripheral sensory neurons in Fabry disease. Histologic and morphometric evaluation of cutaneous nerves, spinal ganglia, and posterior columns. Arch Neurol. 1974;31:120-7.
- 31. Lao LM, Kumakiri M, Mima H, Kuwahara H, Ishida H, Ishiguro K, et al. The ultrastructural characteristics of eccrine sweat glands in Fabry disease patient with hypohidrosis. J Dermatol Sci. 1998;18:109-17
- 32. Cable WJ, Kolodny EH, Adams RD. Fabry disease: impaired autonomic function. Neurology. 1982;32:498-502
- 33. Möhrenschlager M, Braun-Falco M, Ring J, Abeck D. Fabry disease: recognition and management of cutaneous manifestations. Am J Clin Dermatol. 2003;4:189-96
- 34. Lidove O, Ramaswami U, Jaussaud R, Barbey F, Maisonobe T, Caillaud C, et al. Hyperhidrosis: a new and often early symptom in Fabry disease. International experience and data from the Fabry Outcome Survey. Int J Clin Pract. 2006;60:1053-9
- 35. Amann-Vesti BR, Gitzelmann G, Widmer U, Brosshard NU, Steinmann B, Koppensteiner R. Severe lymphatic microangiopathy in Fabry disease. Lymphat Res Biol. 2003;1:185-9
- 36. Nakai K, Yoneda K, Abe T, Moriue T, Matsuoka Y, Nibu N, et al. Multiple leg ulcers in a patient with Fabry disease. J Eur Acad Dermatol Venereol. 2008;22:382-3
- 37. Chen HJ, Yang CC, Hsiao CH, Chu CY. Cutaneous polyarteritis nodosa in a patient with Fabry disease. Arch Dermatol. 2008;144:122-3
- 38. Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ, et al. Pediatric Fabry disease. Pediatrics. 2005;115:344-55
- 39. Birklein F. Mechanisms of neuropathic pain and their importance in Fabry disease. Acta Paediatr Suppl. 2002;91:34-7
- 40. Morgan SH, Rudge P, Smith SJ, Bronstein AM, Kendall BE, Holly E, et al. The neurological complications of Anderson-Fabry disease (alpha-galactosidase A deficiency): investigation of symptomatic and presymptomatic patients. Q J Med. 1990;75:491-507
- 41. Linhart A, Kampmann C, Zamorano JL, Sunder- Plassman G, Beck M, Mehta A, et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28:1228-35
- 42. Sher NA, Letson RD, Desnick RJ. The ocular manifestations in Fabry's disease. Arch Ophthalmol. 1979;97:671-6
- 43. Orssaud C, Dufier J, Germain D. Ocular manifestations in Fabry disease: a survey of 32 hemizygous male patients. Ophthalmic Genet. 2003;24:129-39
- 44. Nguyen TT, Gin T, Nicholls K, Low M, Galanos J, Crawford A. Ophthalmological manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. Clin Experiment Ophthalmol. 2005;33:164-8
- 45. Hirano K, Murata K, Miyagawa A, Terasaki H, Saigusa J, Nagasaka T, et al. Histopathologic findings of cornea verticillata in a woman heterozygous for Fabry's disease. Cornea. 2001;20:233-6
- 46. Desnick RJ, Wasserstein MP, Banikazemi M. Fabry disease (a-galactosidase A deficiency): renal involvement and enzyme replacement therapy. In: Schieppati A, Daina E, Sessa A, Ramuzzi G, editors. Rare Kidney Diseases. Contrib Nephrol. Basel, Switzerland: Karger; 2001. p. 234-40
- 47. Kampmann C, Baehner F, Ries M, Beck M. Cardiac involvement in Anderson-Fabry disease. J Am Soc Nephrol. 2002;13:S147-9
- 48. Linhart A, Palecek T, Bultas J, Ferguson JJ, Hrudová J, Karetová D, et al. New insights in cardiac structural changes in patients with Fabry's disease. Am Heart J. 2000;139:1101-8
- 49. Perrot A, Osterziel KJ, Beck M, Dietz R, Kampmann C. Fabry disease: focus on cardiac manifestations and molecular mechanisms. Herz. 2002;27:699-702
- 50. Cartwright DJ, Cole AL, Cousins AJ, Lee PJ. Raised HDL cholesterol in Fabry disease: response to enzyme replacement therapy. J Inherit Metab Dis. 2004;27:791-3
- 51. Heltianu C, Costache G, Azibi K, Poenaru L, Simionescu M. Endothelial nitric oxide synthase gene polymorphisms in Fabry's disease. Clin Genet. 2002;61:423-9
- 52. Hauser AC, Lorenz M, Sunder-Plassmann G. The expanding clinical spectrum of Anderson-Fabry disease: a challenge to diagnosis in the novel era of enzyme replacement therapy. J Intern Med. 2004;255:629-36
- 53. Ries M, Moore DF, Robinson CJ, Tifft CJ, Rosenbaum KN, Brady RO, et al. Quantitative dysmorphology assessment in Fabry disease. Genet Med. 2006;8:96-101
- 54. Baccaglini L, Schiffmann R, Brennan MT, Lancaster HE Jr, Kulkarni AB, Brahim JS. Oral and craniofacial findings in Fabry's disease: a report of 13 patients. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001;92:415-9
- 55. Möhrenschlager M, Henkel V, Ring J. Angiokeratomas, Fabry disease and enzyme replacement therapy: still a challenge. Br J Dermatol. 2005;152:177-8
- 56. Chamoles NA, Blanco M, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta. 2001;308:195-6
- 57. Pastores GM, Lien YH. Biochemical and molecular genetic basis of Fabry disease. J Am Soc Nephrol. 2002;13:S130-3
- 58. Germain DP. Fabry disease. Clinical and genetic aspects. Therapeutic perspectives. Rev Med Interne. 2000;21:1086-103
- 59. Vedder AC, Strijland A, vd Bergh Weerman MA, Florquin S, Aerts JM, Hollak CE. Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis. 2006;29:106-11
- 60. Navarro C, Teijeira S, Dominguez C, Fernandez JM, Rivas E, Fachal C, et al. Fabry disease: an ultrastructural comparative study of skin in hemizygous and heterozygous patients. Acta Neuropathol. 2006;111:178-85
- 61. Kanekura T, Fukushige T, Kanda A, Tsuyama S, Murata F, Sakuraba H, et al. Immunoelectron-microscopic detection of globotriaosylceramide accumulated in the skin of patients with Fabry disease. Br J Dermatol. 2005;153:544-8
- 62. Cabrera H, García S. Angioqueratomas. In: Cabrera H, García S, editors. Nevos. Buenos Aires, Argentina: Actualizaciones Médicas S.R.L.; 1998. p. 123-6
- 63. Pravatà G, Noto G, Aricò M. Angiocheratoma corporis diffusum with normal enzyme activities. G Ital Dermatol Venereol. 1990;125:401-3
- 64. Bennett RL, Hart KA, O'Rourke E, Barranger JA, Johnson J, MacDermot KD, et al. Fabry disease in genetic counseling practice: recommendations of the National Society of Genetic Counselors. J Genet Couns. 2002;11:121-46
- 65. Breunig F, Weidemann F, Beer M, Eggert A, Krane V, Spindler M, et al. Fabry disease: diagnosis and treatment. Kidney Int Suppl. 2003;63:S181-5
- 66. Warnock DG, West ML. Diagnosis and management of kidney involvement in Fabry disease. Adv Chronic Kidney Dis. 2006;13:138-47
- 67. Reis Pina P. O coração e a doença de Fabry-Anderson. Med Int. 2003;10:209-14
- 68. Desnick RS, Brady R, Barrenger J, Collins AJ, Germain DP, Goldman M, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Int Med. 2003;138:338-46
- 69. Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8:539-48
- 70. Siatskas C, Medin JA. Gene therapy for Fabry disease. J Inherit Metab Dis. 2001;24:S25-41
- 71. Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet. 2002;3:954-66
- 72. Roth J, Yam GH, Fan J, Hirano K, Gaplovska-Kysela K, Le Fourn V, et al. Protein quality control: the who's who, the where's and therapeutic escapes. Histochem Cell Biol. 2008;129:163-77
- 73. Fan JQ, Ishii S, Asano N, Suzuki Y. accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med. 1999;5:112-5
- 74. Yam GH, Bosshard N, Zuber C, Steinmann B, Roth J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. Am J Physiol Cell Physiol. 2006;290:C1076-82
- 75. Abe A, Gregory S, Lee L, Killen PD, Brady RO, Kulkarni A, et al. Reduction of globotriaosylceramide in Fabry disease mice by substrate deprivation. J Clin Invest. 2000;105:1563-71

Datas de Publicação
-
Publicação nesta coleção
19 Out 2009 -
Data do Fascículo
Ago 2009