Abstracts
Tuberous sclerosis is a rare autosomal dominant disorder. Myofibroblasts are cells with a hybrid phenotype between fibroblasts and smooth muscle cells. The objective of this study is to describe clinical and histopathological characteristics of tuberous sclerosis and to conduct an immunohistochemical evaluation of myofibroblasts in cutaneous angiofibromas present in this condition. Lesion sections removed were stained with hematoxylin-eosin and Masson's trichrome. Immunohistochemistry against α-SMA was done to determine the presence of myofibroblasts, and the reaction was negative. Since α-SMA is a specific marker for myofibroblasts, this result suggests that myofibroblasts are not involved in cutaneous angiofibromas present in the tuberous sclerosis case reported.
Angiofibroma; Genetics; Tuberous sclerosis
Esclerose tuberosa é uma condição rara e autossômica dominante. Miofibroblastos são células que exibem fenótipo híbrido entre fibroblastos e células musculares lisas. O objetivo deste caso clínico é mostrar as características clínicas e histopatológicas da esclerose tuberosa e avaliar miofibroblastos nos angiofibromas cutâneos dessa condição. Lesões removidas foram coradas em HE e tricrômico de Masson. Para determinar a presença de miofibroblastos foi usada imunoistoquímica para α-SMA. Essa reação mostrou-se negativa para miofibroblastos. Como α-SMA é um marcador específico para essas células, esse resultado sugere que miofibroblastos não estejam envolvidos com os angiofibromas da esclerose tuberosa descrita.
Angiofibroma; Esclerose tuberosa; Genética
CASE REPORT
IPhysician specialized in oral maxillofacial surgery at Universidade de Alfenas (Unifenas) - Alfenas (MG), Brazil
IIPh.D. student in Stomatopathology at Universidade Estadual de Campinas (Unicamp) - Piracicaba (SP), Brazil
IIIProfessor, Ph.D., at Universidade Estadual de Montes Claros (Unimontes) - Montes Claros (MG), Brazil
IVProfessor of Odontology, Universidade de Alfenas (Unifenas) - Alfenas (MG), Brazil
VProfessor of Medical Sciences, Universidade de Alfenas (Unifenas) - Alfenas (MG), Brazil
VIFull Professor at Universidade Estadual de Montes Claros (Unimontes) - Montes Claros (MG), Brazil
Mailing Address
ABSTRACT
Tuberous sclerosis is a rare autosomal dominant disorder. Myofibroblasts are cells with a hybrid phenotype between fibroblasts and smooth muscle cells. The objective of this study is to describe clinical and histopathological characteristics of tuberous sclerosis and to conduct an immunohistochemical evaluation of myofibroblasts in cutaneous angiofibromas present in this condition. Lesion sections removed were stained with hematoxylin-eosin and Masson's trichrome. Immunohistochemistry against α-SMA was done to determine the presence of myofibroblasts, and the reaction was negative. Since α-SMA is a specific marker for myofibroblasts, this result suggests that myofibroblasts are not involved in cutaneous angiofibromas present in the tuberous sclerosis case reported.
Keywords: Angiofibroma; Genetics; Tuberous sclerosis
INTRODUCTION
Tuberous sclerosis is a congenital autosomal dominant disorder with high penetration and variable expression. It is a condition with universal distribution and no predilection for race or gender.1 Its estimated prevalence is of 1:10.000 live births2; however, in more than 50% of the cases its incidence can range from 1:50.000 to 1:300.000, due to the high frequency of new mutations3. About 50% to 84% of occurrences do not show familial history. 3
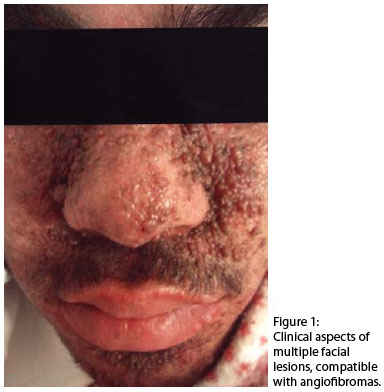
Tuberous sclerosis is a multisystemic genetic disease, characterized in its complex form by lesions that affect the brain, kidneys, lungs, skin, and other organs.4 Mucocutaneous manifestations rarely appear at birth; they develop during puberty1. Classical cutaneous lesions correspond to multiple angiofibromas in the face, which usually appear between 5 years of age and puberty. Angiofibromas often appear in the face, commonly in the form of "butterfly wings" (nasogenian folds) with flaccid appearance and pale pink color.5
Myofibroblasts are mesenquimal cells with hybrid phenotype - they show characteristics of fibroblasts and smooth muscle cells. They are characterized by high expression of ?-actin isoforms (?-SMA).6 Studies have shown the presence of cells with myofibroblastic features in normal specialized tissues and in several abnormal conditions, such as Dupuytren's disease, hereditary gingival fibromatosis, cyclosporine A-induced fibrous pancreatic fibrosis, and in renal, hepatic, and pulmonary fibromatosis.7,8
Hence, the objective of this study is to evaluate the presence of myofibroblasts in cutaneous angiofibromas of tuberous sclerosis.
CASE REPORT
Male patient, with leukoderma, 18 years old, diagnosed with tuberous sclerosis, in a wheelchair. He was referred, accompanied by his mother, to this university hospital for evaluation and treatment, in addition to odontalgia. In anamnesis, it was reported that the patient presented oligophrenia, constipation, and multiple skin lesions. Prior history of epileptic seizures was also reported. The patient was being treated with Clonazepam (6 mg/day), Phenobarbital (300 mg/day), Lamotrigine (200 mg/day), Clobazam (20 mg/day), and Benzathine Penicillin (1 ampoule/21 days). In the clinical examination, extensive folliculitis was observed in the inguinal area and severe acne, in the cervical region and dorsum of thorax, with considerable scarring and fistulous areas. The patient showed multiple lesions (compatible with angiofibromas) asymptomatic and firm on palpation, distributed in the face and thorax, respectively (Figures 1 and 2). In addition to cutaneous lesions, defects in tooth ename were observed, which were compatible with amelogenesis imperfecta. The mother informed that there was no history of consanguinity in the family and that her other children did not present any abnormality. She also said that her son's lesions increased substantially over the past 3 years.
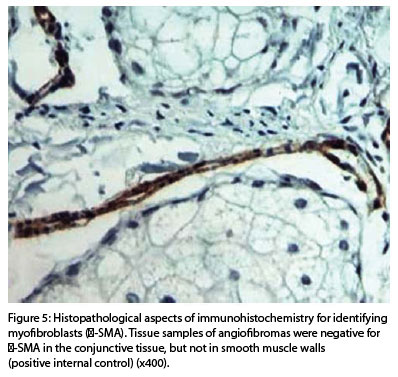
After clinical examination, the patient was submitted to complementary examinations (including imaginologic exams) of the various internal organs normally affected by tuberous sclerosis. Cranial computed tomography revealed images indicative of multiple intracranial calcifications, in addition to cortical sulci atrophy, and third ventricle dilation. Surgical removal of cutaneous lesions was performed under general anesthesia. After removal, lesions were fixed in 10% formol and submitted to histopathological processing. Tissue sections were embedded in paraffin and stained in hematoxylin-eosin (HE) and Masson's trichrome (MT). In the hematoxylin and eosin staining of tissue sections the following were observed: dense conjunctive tissue with an irregular disposition and numerous collagen fibers, blood vessels, mononuclear inflammatory infiltrate, hypermelanotic surface epithelium and annexes. These microscopical characteristics were compatible with the diagnosis of angiofibroma (Figure 3). Masson's trichrome staining showed conjunctive tissue with collagen fibers in blue, blood vessels, and mononuclear inflammatory infiltrate (Figure 4). In order to determine the presence of myofibroblasts in skin lesions, tissues were submitted to immunohistochemical reaction against -SMA proteins.9 Tissue samples of angiofibromas were negative for -SMA in the conjunctive tissue, but not in smooth muscle walls (positive internal control) (Figure 5).
DISCUSSION
Although tuberous sclerosis represents a genetic autosomal dominant condition, the mechanisms involved in this disorder are not fully understood. Genetic alterations were, until recently, described in two distinct loci: TSC1 and TSC210. The TSC1 gene is responsible for 80-85% of mutations. It is located in the long arm of chromosome 9q34 and encodes for the protein hamartin, which regulates biochemical adhesive events.10 The TSC2 gene answers for 15-20% of mutations in tuberous sclerosis and is associated with more severe clinical manifestations.10 It is located in chromosome 16p13 and encodes for the protein tuberin, which regulates vesicular transportation from the cytoplasm to the membrane and the mechanisms of mitotic regulation.11 Both genes show inhibitory effects over cell growth, proliferation, and migration because they stimulate the passage of cells that were latent in the G0 phase, in the cellular cycle, in the S phase. It is also known that hamartin alters cellular adhesion and tuberin activates Ras gene, but the manner in which these two proteins interact, as tumor suppressor genes, is still unknown. Hence, the absence or hypofunction of these tumor suppressive genes is associated with the clinical condition of tuberous sclerosis.10,11
In the present case report, the patient did not have positive familial history of tuberous sclerosis and there was no history of consanguineous marriage between the parents. The patient was the only case in the family, probably due to a new mutation. The mother reported that the patient suffered epileptic seizures from infancy, with neurologic alterations (oligophrenia). Suggestive images of multiple intracranial calcifications, in addition to cortical sulci atrophy, and third ventricle dilation were observed in cranial computed tomography. The most prominent neurologic manifestations of tuberous sclerosis include developmental delay, seizures, and behavioral problems. Tubercles of tuberous sclerosis are focal lesions that predominate in gray matter and are more pale and of firmer consistency than that of the brain.11
Classical cutaneous alterations in tuberous sclerosis patients include multiple facial angiofibromas, usually in nasogenian folds, zygomatic area and chin. Leafy hypomelanotic maculae and periungueal and lumbosacral fibromas are also observed in this dermatologic condition.10 Cutaneous lesions often appear between 5 years of age and puberty. They are small tumors located inside the dermis. Leafy hypomelanotic maculae, confetti cutaneous lesions, Shagreen patches, periungueal fibromas, and fibrotic plaques in the lumbosacral region may be present. 11 Other lesions that occur in tuberous sclerosis are conjunctive tissue hamartomas, which can be vascular, fibrous or dermal. Multiple periungueal fibromas or Koenen's tumors are pink formations that originate in the corner of nails of the foot and histologically resemble neurofibromas.11 The patient presented multiple cutaneous lesions with histopathological diagnosis of angiofibroma. The mother reported that the lesions appeared in infancy and had become worse in the last three years, becoming more concentrated in the face.
Histopathological aspects found in HE and MT staining were similar to those described in the literature, compatible with the microscopical diagnosis of angiofibroma. After analysis in HE and MT staining, lesions were submitted to immunohistochemical reactions to determine the presence of myofibroblasts. In the present clinical case myofibroblasts were not observed. These cells, when activated, synthesize high levels of protein from the extracellular matrix, especially collagen, and reduced levels of metalloproteinase from the matrix, participating in various fibrotic processes. Morphologically, myofibroblasts are characterized by the presence of a contractile apparatus that contains bundles of actin microfilaments, especially ?-SMA, associated with proteins such as myosin and stress fibers.12 The mechanisms that give origin to the transdifferentiation of myofibroblasts remain unknown; however, the participation of TGF-β1 in this process has been demonstrated. 13 Blocking methods of the synthesis of TGF-β1 have been tested as treatment for diseases caused by interstitial fibrosis.13
The criteria for diagnosis of tuberous sclerosis were suggested by Gomez (1979)14, who divided them into major and minor. This classification system is based on clinical and imaginologic aspects. However, the classical triad of the disorder consists of epilepsy, developmental delay, and facial angiofibromatosis.11 In the present case, the patient showed characteristics for the clinical diagnosis of tuberous sclerosis. In addition to dermatologic and neurological characteristics, dental alterations were also observed, which were compatible with amelogenesis imperfecta. The patient has been receiving follow-up care for a year, with no signs of lesion recidivism so far. In addition to dermatologic treatment, the patient received odontological treatment, and his mother received orientation for adequate mouth hygiene.
REFERENCES
- 1. Jó_wiak S, Schwartz RA, Janniger CK, Micha_owicz R, Chmielik J. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course and diagnostic significance. Int J Dermatol. 1998;37:911-7.
- 2. Louis DN, von Deimling A. Hereditary tumor syndromes of the nervous system: overview and rare syndromes. Brain Pathol. 1995;5:145-51.
- 3. Kandt RS. Tuberous sclerosis complex and neurofibromatosis type 1: the two most common neurocutaneous diseases. Neurol Clin. 2002;20:941-64.
- 4. MacCollin M, Kwiatkowski D. Molecular genetic aspects of the phakomatoses: tuberous sclerosis complex and neurofibromatosis 1. Curr Opin Neurol. 2001;14:163-9.
- 5. Short MP, Adams RD. Neurocutaneous diseases. In: Fitzpatrick T, Eisen AZ, Wolff K, Freedberg IM, Austen KR, editors. Dermatology in general medicine. 4th ed. New York: McGraw-Hill;1993. p. 2249-90.
- 6. Gabbiani G. The biology of the myofibroblasts. Kidney International. 1992;41:530-2.
- 7. Bitu CC, Sobral LM, Kelemann MG, Martelli-Júnior H, Zecckin KG, Graner E, et al. Heterogeneous presence of myofibroblasts in hereditary gingival fibromatosis. J Clin Periodontol. 2006;33:393-400.
- 8. Willis BC, duBois RM, Borok Z. Ephitelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377-82.
- 9. Bozzo L, Machado MAN, Almeida OP, Lopes MA, Coletta RD. Hereditary gingival fibromatosis: report of three cases. J Clin Pediatr Dent. 2000; 25:41-6.
- 10. Almeida-Jr HL. Dermatologia comparative. An Bras Dermatol. 2005;80:307-8.
- 11. Berbet ALCV, Fernandes CAA. Esclerose tuberosa. An Bras Dermatol. 1997;72:73-7.
- 12. Desmouliére A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and myofibroblasts. Wound Repair Regen. 2005;13:7-12.
- 13. Smith PC, Cáceres M, Martinez J. Induction of the myofibroblastic phenotype in human gingival fibroblasts by transforming growth factor beta: role of Rhoa-Rock and c-Jun N-terminal kinase signaling pathways. J Periodontal Res. 2006;41:418-25.
- 14. Gomez MR. Tuberous sclerosis. New York: Raven Press;1979.
Tuberous sclerosis: evaluation of myofibroblasts in cutaneous angiofibromas - case report
Publication Dates
-
Publication in this collection
06 May 2010 -
Date of issue
Feb 2010
History
-
Received
16 Mar 2009 -
Accepted
30 June 2009