Resumos
A síndrome de Bart é caracterizada pela tríade de manifestações clínicas: epidermólise bolhosa, aplasia de cútis e anormalidades ungueais. Presume-se que a ausência congênita de pele segue as linhas de Blaschko. O tratamento visa a evitar infecções secundárias. O prognóstico é bom e depende da cicatrização das bolhas.
Anomalias congênitas; Epidermólise bolhosa; Genética
Bart's syndrome is characterized by three clinical manifestations: epidermolysis bullosa, congenital absence of skin, and nail abnormalities. It is believed that congenital skin absence follows the patterns of Blaschko's lines. Treatment seeks to prevent secondary infection. Prognosis is favorable and depends upon the healing of lesions.
Congenital abnormalities; Epidermolysis bullosa; Genetics
SÍNDROME EM QUESTÃO
Você conhece esta síndrome?
Nadia Aparecida Pereira de AlmeidaI; Flavia SerafiniII; Janaine MarchioriIII; Julie Gomes Del MoroIV
IMédica dermatologista responsável pelo serviço de dermatologia pediátrica do Hospital Infantil Pequeno Príncipe - Curitiba (PR), Brasil
IIMédica residente de dermatologia do Hospital de Clínicas - Curitiba (PR), Brasil
IIIAcadêmica de medicina da Pontifícia Universidade Católica do Paraná - Curitiba (PR), Brasil
IVAcadêmica de medicina da Pontifícia Universidade Católica do Paraná - Curitiba (PR), Brasil
Endereço para correspondência Endereço para correspondência: Janaine Marchiori Rua João Florindo Zanetti, 3.310 83606 200 Campo Largo, PR
RESUMO
A síndrome de Bart é caracterizada pela tríade de manifestações clínicas: epidermólise bolhosa, aplasia de cútis e anormalidades ungueais. Presume-se que a ausência congênita de pele segue as linhas de Blaschko. O tratamento visa a evitar infecções secundárias. O prognóstico é bom e depende da cicatrização das bolhas.
Palavras-chave: Anomalias congênitas; Epidermólise bolhosa; Genética
RELATO DO CASO
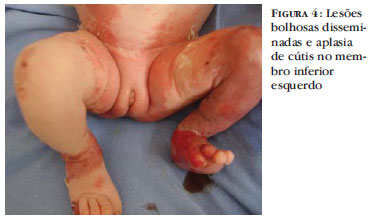
Paciente ABA, feminina, branca, natural e procedente de Piraí do Sul, Paraná. Mãe de 18 anos, primigesta e sem intercorrências durante o pré-natal. Nasceu de parto vaginal, a termo, Apgar 9/10, pesando 2.205 gramas. Os pais são saudáveis e primos em primeiro grau. Foi encaminhada por lesões bolhosas disseminadas, incluindo mucosa oral (Figuras 1 e 2) e extensa área de ausência de pele no membro inferior esquerdo, desde o joelho até o pé (Figura 2). Apresentava também anoníquia no primeiro quirodáctilo direito e no primeiro e segundo pododáctilos esquerdos (Figura 3). Submetida a biopsia de pele, cujo padrão histológico foi compatível com epidermólise bolhosa. Durante o internamento necessitou de transfusão de concentrado de hemácias. Recebeu oxacilina por 10 dias e amicacina por cinco dias. Foram realizados curativos diários com ácido fusídico, adaptic®, loção oleosa à base de ácidos graxos essenciais e creme de nitrato de cério 12,4% com sulfadiazina de prata 1%. Na alta as lesões bolhosas estavam em processo de cicatrização (Figura 4).
QUE SÍNDROME É ESTA?
Atualmente considerada uma doença genética gênica por Christiano e col., a síndrome de Bart é caracterizada pela tríade de manifestações clínicas: ausência congênita localizada de pele; bolhas, pústulas, vesículas mucocutâneas (epidermólise bolhosa) e alterações ungueais.
Uma das explicações sugeridas por vários autores para a ausência congênita de pele é uma lesão mecânica causada por trauma físico intraútero. Outros autores suportam a ideia de que isso seria plausível para lesões unilaterais, pois lesões de membros inferiores bilaterais e simétricas, principalmente em pododáctilos e planta dos pés, dificilmente surgiriam por fricção. McKinster e col. apoiam-se na ideia de que a área acometida segue as linhas de Blaschko, as quais são segmentos metaméricos como dermátomos que direcionam o crescimento de clones de células cutâneas que derivam de um número limitado de precursores. As lesões desnudas que seguem tais linhas são simétricas, com bandas largas em formato de "S" e com bordas definidas.1
As alterações ungueais encontradas nessa síndrome são distrofias ungueais, onicomadese e anoníquia.1,2 Em relação às lesões bolhosas mucocutâneas, considera-se ser a síndrome de Bart uma variante intraútero da epidermólise bolhosa distrófica. Essa afirmação é embasada na descoberta de uma alteração no braço curto do cromossomo 3 na região p21 próximo da região do gene do colágeno tipo VII COL7A1, com substituição de glicina por arginina, alteração esta encontrada tanto na síndrome de Bart quanto em outras variantes de epidermólise bolhosa distrófica.2 Outra hipótese dessa afirmação é que a ultraestrutura revela vesículas na sublâmina densa, queratinócitos basais, tonofilamentos e hemidesmossomos normais e fibrilas de ancoramento pobremente formadas ou em número diminuído. As diferenças fenotípicas entre as duas patologias citadas anteriormente resultam de uma substituição da glicina em uma região localizada e específica. Já para Fine e col., a síndrome de Bart simplesmente descreve a ocorrência simultânea de ausência congênita localizada de pele e epidermólise bolhosa congênita e não significa que aquela é uma manifestação da epidermólise bolhosa distrófica.
Apesar de a base molecular não ser clara, classifica-se a síndrome de Bart como uma herança autossômica dominante de penetrância total, mas com expressividade variável.
O tratamento é realizado com antimicrobianos tópicos e gazes estéreis para profilaxia de infecções secundárias, e em poucas semanas as lesões estão suavemente cicatrizadas.1 Outras alternativas são o creme de sulfadiazina de prata em maiores de dois meses de idade e desbridamento precoce das lesões com curativos fechados, que são essenciais para a sobrevida.3 O tratamento das lesões é de suma importância devido ao quadro extremamente doloroso e limitante à qualidade de vida.4
As complicações de um tratamento conservador podem ser sepse, infecção local, hemorragia, excessiva perda de fluido, hipotermia, distúrbios de eletrólitos e, mais tarde, cicatrizes hipertróficas ou atróficas.3 Décadas atrás, quando não havia uma variabilidade de produtos sofisticados e técnica adequada para o cuidado das feridas, a infecção era a maior causa de mortalidade em qualquer um dos tipos de epidermólise bolhosa congênita.5
Atualmente, com a disponibilidade de novos produtos no mercado, o prognóstico é relativamente bom e depende da melhora das bolhas com o passar da idade.

Aprovado pelo Conselho Editorial e aceito para publicação em 27.11.2009.
Suporte financeiro: Nenhum
Conflito de interesse: Nenhum
* Trabalho realizado no Hospital Pequeno Príncipe - Curitiba (PR), Brasil.
- 1. Duran-McKinster C, Rivera-Franco A, Tamayo L, de la Luz Orozco-Covarrubias M, Ruiz-Maldonado R. Bart syndrome: the congenital localized absence of skin may follow the lines of Blaschko. Report of six cases. Pediatr Dermatol. 2000;17:179-82.
- 2. Christiano AM, Bart BJ, Epstein EH, Uitto J. Genetic basis of Bart´s Syndrome: a glycine substitution mutation in the type VII collagen gene. The Society for Investigative Dermatology, Inc. 1996;106:1340-2.
- 3. Ahcan U, Janezic T. Management of aplasia cutis congenita in a non-scalp location. Br J Plast Surg. 2002;55:530-2.
- 4. Horn HM, Tidman MJ. The clinical spectrum of epidermolysis bullosa simplex. Br J Dermatol. 2000;142:468-72.
- 5. Fine JD, Johnson LB, Weiner M, Suchindran C. Cause-specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr. 2008;152:276-80.
Endereço para correspondência:
Datas de Publicação
-
Publicação nesta coleção
06 Maio 2010 -
Data do Fascículo
Fev 2010