Abstracts
Hay-Wells syndrome is a rare form of ectodermal dysplasia initially described by Hay and Wells in 1976. It is an autosomal dominant disorder with varying forms of expression featuring congenital abnormalities of the skin, hair, teeth, nails and sweat glands. The present report describes the case of a 17-yearold white boy, the son of nonconsanguineous parents, who presented ankyloblepharon filiforme adnatum, ectodermal dysplasia and a cleft palate at birth, which are considered cardinal signs of this syndrome by most authors. We also highlight the importance of implementing multidisciplinary follow-up of these patients.
Ectodermal dysplasia; Cleft palate; Syndrome
A síndrome de Hay-Wells é uma forma rara de displasia ectodérmica, descrita inicialmente em 1976 por Hay e Wells, de caráter autossômico dominante com expressão variável, composta por anomalias congênitas da pele, cabelos, dentes, unhas e glândulas sudoríparas. Descrevemos o caso de um paciente de 17 anos, filho de pais não consangüíneos, que apresentava anquiloblefaron filiforme adenatum, displasia ectodérmica e fenda palatina ao nascimento, sinais considerados cardinais pela maioria dos autores. Destacamos também a importância do acompanhamento multidiscliplinar dos pacientes.
Displasia ectodérmica; Fissura palatina; Síndrome
CASE REPORT
Hay-Wells syndrome - A case report
Dário Júnior de Freitas RosaI; Ronaldo Figueiredo MachadoI; Marcelino Pereira Martins NetoII; Alessandra Almeida Montenegro de SáII; Aloísio GamonalIII
IResident Physician in Dermatology, Federal University of Juiz de Fora (UFJF), Juiz de Fora, MG, Brazil
IIDermatologist, Preceptor, Federal University of Juiz de Fora (UFJF), Juiz de Fora, MG, Brazil
IIIProfessor, PhD, Head of the Department of Dermatology, Federal University of Juiz de Fora (UFJF), Juiz de Fora, MG, Brazil
Mailing Address
ABSTRACT
Hay-Wells syndrome is a rare form of ectodermal dysplasia initially described by Hay and Wells in 1976. It is an autosomal dominant disorder with varying forms of expression featuring congenital abnormalities of the skin, hair, teeth, nails and sweat glands. The present report describes the case of a 17-yearold white boy, the son of nonconsanguineous parents, who presented ankyloblepharon filiforme adnatum, ectodermal dysplasia and a cleft palate at birth, which are considered cardinal signs of this syndrome by most authors. We also highlight the importance of implementing multidisciplinary follow-up of these patients.
Keywords: Cleft palate; Ectodermal dysplasia; Syndrome
INTRODUCTION
Hay-Wells syndrome, also known as AEC syndrome (ankyloblepharon-ectodermal dysplasia-clefting syndrome), is a rare genetic disorder, initially described by Hay and Wells1 in 1976 in seven individuals from 4 families in whom multiple and complex malformations were associated with a pattern of autosomal dominant inheritance of varying degrees of penetrance. Sporadic cases have, however, also been described. 2 Later studies showed that this pathology is the result of a mutation in the p63 gene, a homologue of the p53 tumor suppressor gene (TP53).3
Most authors consider the presence of ankyloblepharon filiforme adnatum (tissue strands joining the upper and lower eyelid margins), cleft lip and/or palate and findings consistent with ectodermal dysplasia to constitute essential criteria for a diagnosis of Hay-Wells syndrome. Common ectodermal defects include alopecia, onychodystrophy, hypodontia and hypohidrosis.
CASE REPORT
The present case report describes a white male patient of 17 years of age, an agricultural worker referred to the Dermatology Clinic at the Teaching Hospital of the Federal University of Juiz de Fora complaining of having no fingerprints, a problem that was hampering his attempts to obtain identification documents. He reported constantly watering eyes and heat intolerance. Erythroderma was present at birth, with blistered lesions on his scalp and body, as well as ankyloblepharon filiforme adnatum, reverted by the attending physician at delivery, and a cleft palate repaired by surgery performed when he was nine years old. His hair only began to grow when he was three years old and his teeth at five years of age. The patient also reported multiple scalp infections treated intermittently with varying courses of oral antibiotics since infancy.
He was born to nonconsanguineous parents and there were no similar cases in the family.
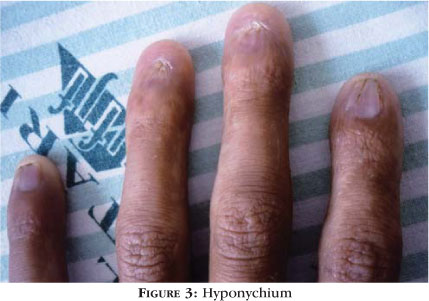
Dermatological evaluation revealed sparse, fair hair with areas of alopecia, hypertelorism, a lower lip that was more prominent than the upper (Figure 1), oligodontia, an overall sparseness of body hair and reticulated hyperpigmentation of the skin, principally on the neck, the upper portion of the trunk and proximal limbs (Figure 2). Evaluation of the extremities revealed hyponychium and punctate palmoplantar keratoderma (Figures 3 and 4).
The patient had no intellectual impairment. His weight and height were normal for his age.
Laboratory tests including complete blood count, electrolytes and liver, kidney and thyroid function tests showed no abnormalities. Chest radiography was normal.
Histopathology of a fragment of skin from the scalp showed an epidermis with no abnormalities and a dermis with a reduced number of skin adnexa, only one mature pilous follicle being found, as well as some sebaceous glands and mild lymphocytic inflammatory infiltrate.
DISCUSSION
Hay-Wells or AEC syndrome is a rare, autosomal dominant disease. Ankyloblepharon filiforme adnatum, ectodermal dysplasia and cleft lip and/or palate are considered cardinal signs of this syndrome by most authors.4 All these features were present in the case reported here.
Ankyloblepharon consists of partial or complete fusion of the upper and lower eyelid margins, and is often sporadic. Normally, the eyelids remain fused until the fifth week of pregnancy at which time they separate. Therefore, anomalies that occur between the 7th and 15th weeks of pregnancy may result in palpebral abnormalities.5 Ankyloblepharon may also be present in trisomy 18 and CHAND (Curly Hair-Ankyloblepharon-Nail Dystrophy) syndrome, and is associated with cardiac defects, hydrocephaly, imperforate anus and glaucoma; therefore, its presence should always serve as an alert to the possibility of another important concomitant disorder.6
Ectodermal dysplasias refers to a group of diseases in which defects occur in the development of the hair, teeth, nails, sweat glands and other structures originating from the ectoderm.7,8 These abnormalities, when associated with other malformations, constitute a group of ectodermal dysplasia syndromes that includes the ectrodactyly-ectodermal dysplasiaclefting (EEC) syndrome, Rapp-Hodgkin syndrome and the CHAND syndrome, which represent the principal differential diagnoses for the presently reported syndrome.8
Patients with Hay-Wells syndrome may present various degrees of alopecia, sparse, fair hair, onychodystrophies, palmoplantar hyperkeratosis, skin pigmentation disorder,9 hypohidrosis, hypodontia, dental malformations and auricular deformities,10 all of which were present in the patient in question. Lacrimal duct obstruction is a common finding. Other reported findings include supernumerary nipples, otitis media, hypospadia, midfacial hypoplasia, hypertelorism, low stature, intellectual impairment, hypoacousia and ocular abnormalities.
At birth, the child may present erythroderma with desquamation, superficial erosion and crusting. On the scalp, erosive dermatitis is present, and is a common source of secondary infection that puts these patients at a greater risk of bacterial superinfection and sepsis, thereby leading to a considerable increase in the morbidity and mortality of neonates with this pathology.11 Various reports have been published describing newborn infants with Hay-Wells syndrome who were erroneously diagnosed with epidermolysis bullosa due to the presence of erythroderma and extensive areas of erosion. In association with the classic characteristics of this syndrome, skin erosion in the newborn infant and the recurrent scalp infection, as found in the patient in the present report, are important signs that aid in the differential diagnosis with the other forms of ectodermal dysplasia.2,7 It is suggested that the scalp lesions tend to resolve with age and/or progress with alopecia.2
This syndrome is caused by a mutation in the p63 gene, a homologue of the p53 tumor suppressor gene (TP53),2,3 which plays a role in the process of stratifying the epidermal epithelium, regulating the proliferative capacity of the basal keratinocytes. The evidence that an alteration in that gene may result in other pathologies such as the EEC and Rapp-Hodgkin syndromes suggests a highly pleomorphic effect of mutations in the p63 gene, the abnormalities associated with Hay-Wells syndrome originating specifically from the substitution of amino acids in the SAM (sterile alpha motif) domain.3,12
Histopathology of skin samples from patients has shown an atrophic epidermis with reduced spinous and granular layers, as well as confluent parakeratosis. 11 The number of scalp follicles is decreased, while the sebaceous glands are decreased in number and size,7,11 although cases have been reported in which no alterations in adnexal structures were found. 2 In the patient in the present case report, a significant overall reduction in skin adnexa was found in the scalp.
The importance of the early diagnosis of this syndrome should be emphasized in order to implement appropriate genetic counselling for parents, as well as clinical and dermatological treatment of the patient at the initial phase of erythroderma and management of scalp infections. In addition, ophthalmological and odontological care should be initiated and cleft lips and palates corrected surgically.
REFERENCES
- 1. Hay RJ, Wells RS. The syndrome of ankyloblepharon, ectodermal defects, and cleft lip and palate: an autosomal dominant condition. Br J Dermatol. 1976;94:277-89.
- 2. Payne AS, Yan AC, Ilyas E, Li W, Seykora JT, Young TL, et al. Two novel TP63 mutations associated with the ankyloblepharon, ectodermal defects, and cleft lip and palate syndrome - a skin fragility phenotype. Arch Dermatol. 2005;141:1567-73.
- 3. McGrath JÁ, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001;10:221-9.
- 4. Macias E, de Carlos F, Cobo J. Hay-Wells syndrome (AEC): a case report. Oral Dis. 2006;12:506-8.
- 5. Mohamed YH, Gong H, Amenyra T. Role of apoptosis in eyelid development. Exp Eye Res. 2003;76:115-23.
- 6. Sharkey D, Marlow N, Stokes J. Ankyloblepharon filiforme adenatum. J Pediatr. 2008;152:594.
- 7. Tsutsui K, Asai Y, Fujimoto A, Yamamoto M, Kubo M, Hatta N. A novel p63 sterile alpha motif (SAM) domain mutation in a Japanese patient with ankyloblepharon, ectodermal defects and cleft lip and palate (AEC) syndrome without ankyloblepharon. Br J Dermatol. 2003;149:395-9.
- 8. Pinheiro M, Freire-Maia N. Ectodermal dysplasias: a clinical classification and a causal review. Am J Med Genet. 1994;53:153-62.
- 9. Drut R, Pollono D, Drut RM. Bilateral nephroblastoma in familiar Hay-Wells syndrome associated with familial reticulate pigmentation of the skin. Am J Med Genet. 2002;110:164-9.
- 10. Kulkarni ML, Deshmukh S, Matani D, Gayatri K. Hay- Wells syndrome of ectodermal dysplasia. Indian J Pediatr.2006;73:101.
- 11. Yoo J, Berk DR, Fabre E, Lind AC, Mallory SB. Ankyloblepharon-ectodermal dysplasia-clefting (AEC) syndrome with neonatal erythroderma: report of two cases. Int J Dermatol. 2007;46:1196-7.
- 12. Bertola DR, Kim CA, Sugayama SM, Albano LM, Utagawa CY, Gonzalez CH. AEC syndrome and CHAND syndrome: further evidence of clinical overlapping in the ectodermal dysplasias. Pediatr Dermatol. 2000;17:218-21.
Publication Dates
-
Publication in this collection
23 June 2010 -
Date of issue
Apr 2010
History
-
Received
27 Apr 2009 -
Accepted
08 May 2009