Abstracts
Ectrodactyly - ectodermal dysplasia - cleft lip/palate syndrome (EEC) is a rare autosomal dominant genetic disorder, with variable expression and penetrance. This congenital disorder is associated either with a mutation in chromosome 7 or with a translocation between chromosomes 7 and 9, reflected primarily in the abnormalities listed in its name. This case report describes a 35-year-old male with syndromic stigmata since birth and no cleft lip/palate. Four relatives are also affected by the condition.
cleft lip; cleft palate; ectodermal dysplasia; syndrome
A síndrome de ectrodactilia, displasia ectodérmica e fenda lábio-palatina (EEC) corresponde a uma rara anomalia genética congênita, de herança autossômica dominante, penetrância e expressividade variáveis, associada à mutação no cromossomo 7 ou translocação entre cromossomos 7 e 9, determinada essencialmente pelas características que a denominam. Relata-se caso de paciente de 35 anos, acometido por estigmas sindrômicos, desde o nascimento, com história familiar e sem fenda lábio-palatina.
Displasia ectodérmica; Fenda labial; Fissura palatina; Síndrome
SYNDROME IN QUESTION
IDermatologist at the Dermatology Department of the Clementino Fraga Filho Teaching Hospital (HUCFF), Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
IIUndergraduate medical student, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
IIIUndergraduate medical student, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
IVUndergraduate medical student, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
VSpecialist in Clinical Genetics, Martagão Gesteira Institute of Child Development and Pediatrics, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
Mailing address
ABSTRACT
Ectrodactyly - ectodermal dysplasia - cleft lip/palate syndrome (EEC) is a rare autosomal dominant genetic disorder, with variable expression and penetrance. This congenital disorder is associated either with a mutation in chromosome 7 or with a translocation between chromosomes 7 and 9, reflected primarily in the abnormalities listed in its name. This case report describes a 35-year-old male with syndromic stigmata since birth and no cleft lip/palate. Four relatives are also affected by the condition.
Keywords: cleft lip; cleft palate; ectodermal dysplasia; syndrome
CASE REPORT
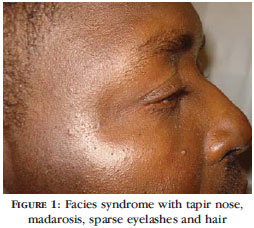
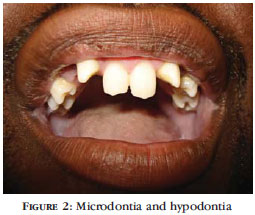
A 35-year old, single, black, male patient from Rio de Janeiro, who was employed as a refrigeration technician, attended a dermatological consultation. Clinical examination revealed facies syndrome with a nose that was broad at the base and collapsed at the point, madarosis, sparse eyelashes, fine, sparse hair (Figure 1), red eyes, hypoplastic and conic teeth (Figure 2) and maxillary hypoplasia, as well as ectrodactyly of the feet and hands (lobster-claw hand) with only two digits (Figures 3 and 4), xerodermia and nail dystrophy (Figure 3). All the photographs shown in the present report were taken with the patient's written consent. At directed anamnesis, the patient denied any other abnormalities in any other parts of the body or systems. He related a family history in which his mother, two siblings and his maternal grandfather all have a similar condition of ectrodactyly in the hands and feet. Ophthalmological examination revealed xerophthalmia, intense blepharitis and chronic ocular hyperemia. He was evaluated by the Department of Clinical Genetics, which confirmed our initial impression of a diagnosis of ectrodactyly - ectodermal dysplasia - cleft lip/palate (EEC) syndrome. Radiography of the patient's hands (Figure 5) and feet correlated with clinical findings, while ultrasonography of the abdomen and urinary tract showed no malformations. The patient is currently being followedup by a multidisciplinary team.
WHAT SYNDROME IS THIS?
Ectrodactyly - ectodermal dysplasia - cleft lip/palate (EEC) syndrome.
EEC syndrome is a rare, autosomal dominant genodermatosis. 2 Around two hundred cases of this syndrome have been described. It results from a mutation in the codifying gene of a tumor suppressor protein (p63) related to the ectoderm and mesoderm. Qumsiyeh suggested that the syndrome originates in chromosome 7 (locus 7q11.2-q21.3), whereas Hasegawa et al. reported cases of reciprocal transposition between chromosomes 7 and 9 (7q11.21 and 9p12 or 7p11.2 and 9q12).3 Cases may occur in families or alone, these single cases generally being the most severe. 3 Interfamilial variability appears to be significantly greater than intrafamilial variability, indicating genetic heterogeneity. 3 The syndrome is characterized by various clinical manifestations that vary significantly between affected individuals, generating different combinations (Table 1). These manifestations include: ectodermal dysplasia such as anatomical or functional abnormalities of the sweat glands 4, fine, sparse, light-colored hair, nail dystrophy; hypoor anodontia; complete or partial cleft lip-palate; abnormalities in tear ducts and eyes; facial hypoplasia; congenital absence of one or more digits (ectrodactyly) with or without syndactyly and clinodactyly; otological abnormalities; reduced stature; genitourinary malformations; impairment of the central nervous system including hearing loss and/or mental retardation; diffuse skin hypopigmentation and cell nevi. 5 In view of the clinical exuberance of this syndrome, there is usually no need for genetic confirmation. Clinical follow-up should be multidisciplinary and the team should include specialists in dermatology, plastic surgery, ophthalmology, nephrology, odontology, clinical genetics, psychology and other specialities, as required. In the majority of cases, surgical intervention of ectrodactyly is unnecessary, since there is no possibility of completely reversing functional damage; nevertheless, it may help with psychosocial aspects. Due to the presence of EEC syndrome in four of the patient's immediate family members, it was not the syndromic stigmata that motivated the patient to seek medical care, leading us to question to what extent his phenotypical abnormalities affect his professional and social activities. The patient in this case report had an incomplete form of EEC syndrome, since cleft lippalate was absent. Other familial cases with ectrodactyly and ectodermal dysplasia without cleft lip/palate have already been reported in the literature, and it has even been suggested that this form of clinical presentation may represent a different nosological entity and not merely a variant of EEC syndrome. 6 The intrafamilial phenotypical variation in this case shows the variability of the penetrance and genetic expression of the syndrome. Differential diagnoses include Goltz syndrome 7, Rapp-Hodgkin syndrome 8 and Hay-Wells syndrome (ankyloblepharon, ectodermal dysplasia and cleft lip and palate syndrome - AEC) 8, all of them being inherited autosomal dominant disorders, including Christ-Siemens-Touraine syndrome 9, a form of hypohidrotic ectodermal dysplasia. Goltz syndrome, which may be an X-linked disorder, is generally lethal in males and is characterized by focal dermal hypoplasia with herniations of adipose tissue, poikiloderma, linear distribution following the lines of Blaschko, abnormalities in body hair, nails, teeth and bones, with ectrodactyly, syndactyly and polydactyly. Rapp-Hodgkin syndrome is characterized by ectodermal dysplasia associated with a narrow nose, cleft lip-palate, microstomia, reduced stature, a prominent forehead, maxillary hypoplasia, a high-arched palate, thin lips and nail hypoplasia. Hay-Wells syndrome is characterized by ectodermal dysplasia, cleft lip/palate and ankyloblepharon.
REFERENCES
- 1. Rüdiger RA, Haase W, Passarge E. Association of ectrodactyly, ectodermal dysplasia, and cleft lip-palate. Am J Dis Child. 1970;120:160-3.
- 2. Brill CB, Hsu LY, Hirschhorn K. The syndrome of ectrodactyly, ectodermal dysplasia and cleft lip and palate: report of a family demonstrating a dominant inheritance pattern. Clin Genet. 1972;3:295-302.
- 3. Odontologia.com. [homepage]. Neves MIR, Lopes FF, Sauáia TS. Síndrome EEC (ectrodactilia, displasia ectodérmica e fenda labial/palatine): relato de caso clínico. Odontologia.com.br. 2004 Abril [acesso 26 Mar. 2009.]. Disponível em: http://www.odontologia.com.br/artigos.asp?id=457
- 4. Peryassú D, Gabriela L. Estudo dos anexos glandulares nas genotermatoses. An Bras Dematol. 1965;40:15-22.
- 5. Almeida SFF, Solari HP. Displasia ectodérmica, ectrodactilia e fissura lábio-palatal: manifestações oculares da síndrome em relato de caso. Arq Bras Oftalmol. 2007;70:125-8.
- 6. Thakkar S, Marfatia Y. EEC syndrome sans clefting: varaiable clinical presentations in a family. Indian J Dermatol Venereol Leprol. 2007;73:46-8.
- 7. Larralde M, Boggio P. Outras genodermatoses. In: Ramos-e-Silva M, Castro MCR. Fundamentos de dermatologia. Rio de Janeiro: Editora Atheneu; 2009. p.289-91.
- 8. Cyriac MJ, Lapashpa E. Lobster-claw hand: a manifestation of EEC syndrome. Indian J Dermatol Venereol Leprol. 2006;72:54-6.
- 9. Succi IB, Fontenelle E. Caso para diagnóstico. Displasia ectodérmica: síndrome de Christ-Siemens-Touraine. An Bras Dermatol. 2009;84:194-6.
Do you know this syndrome?
Publication Dates
-
Publication in this collection
01 Oct 2010 -
Date of issue
Aug 2010
History
-
Received
27 Nov 2009