Resumos
A Síndrome de Huntchinson-Gilford (Progeria) é uma rara doença autossômica dominante, caracterizada pelo envelhecimento precoce. Relata-se caso de uma criança, que aos 6 meses iniciou alopecia na região occipital e placas esclerodermiformes no abdome. Esta síndrome apresenta alterações em vários órgãos e sistemas como a pele, esquelético e sistema cardiovascular. O diagnóstico é clínico e não possui tratamento, porém seu reconhecimento é necessário para minimizar a aterosclerose precoce através do controle da dislipidemia.
Diagnóstico; Envelhecimento; Progeria; Senilidade prematura
Huntchinson-Gilford Syndrome (Progeria) is a rare autosomal dominant disease characterized by premature aging. It is reported the case of child whose alopecia started at the age of 6 months on the occipital region. The child also presented scleroderma plaques on the abdomen. This syndrome presents alterations in many organs and systems such as the skin and the skeletal and cardiovascular systems. The diagnosis is clinical and there is no treatment for it but recognition is necessary to minimize early atherosclerosis through the control of dyslipidemia.
Aging; Aging, premature; Diagnosis; Progeria
SÍNDROME EM QUESTÃO
Você conhece esta síndrome?* * Trabalho realizado na Fundação Alfredo da Matta - Manaus (AM), Brasil.
Livia Lima de LimaI; Carla Barros da Rocha RibasII; Priscilla Maria Rodrigues PereiraIII; Renata Almeida SchettiniI; Josie da Costa EirasIV
IMédica Residente em Dermatologia pela Fundação Alfredo da Matta - Manaus (AM), Brasil. Médica Residente em Dermatologia pela Fundação Alfredo da Matta -Manaus (AM), Brasil
IIMestre em Patologia Tropical pela Universidade Federal do Amazonas (UFAM) - Médica Dermatologista pela Fundação Alfredo da Matta - Manaus (AM) Preceptora da Residência Médica em Dermatologia pela Fundação Alfredoda Matta - Manaus (AM), Brasil
IIIMédica Residente em Dermatologia pela Universidade Federal do Amazonas (UFAM) - Manaus (AM), Brasil. - Médica Residente em Dermatologia pela Universidade Federal do Amazonas (UFAM) - Manaus (AM), Brasil
IVMédica Dermatologista do Hospital Geral de Belém - Exército Brasileiro - Belém (PA), Brasil
Endereço para correspondência Endereço para correspondência: Livia Lima de Lima Rua A29 Conjunto Ajuricaba - 293, Planalto 69046310 Manaus - AM, Brasil Celular: 92 9985 5252 Email: lilima_nb@hotmail.com
RESUMO
A Síndrome de Huntchinson-Gilford (Progeria) é uma rara doença autossômica dominante, caracterizada pelo envelhecimento precoce. Relata-se caso de uma criança, que aos 6 meses iniciou alopecia na região occipital e placas esclerodermiformes no abdome. Esta síndrome apresenta alterações em vários órgãos e sistemas como a pele, esquelético e sistema cardiovascular. O diagnóstico é clínico e não possui tratamento, porém seu reconhecimento é necessário para minimizar a aterosclerose precoce através do controle da dislipidemia.
Palavras-chave: Diagnóstico; Envelhecimento; Progeria; Senilidade prematura
RELATO DO CASO
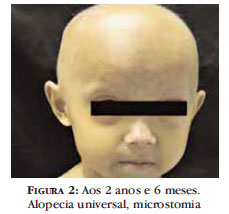
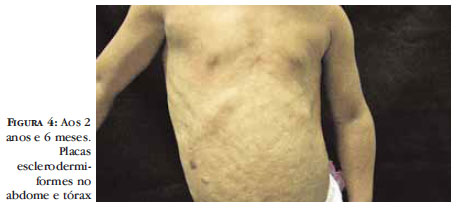
Paciente feminino, com 1 ano e 4 meses, apresentava desde 6 meses de idade perda progressiva dos cabelos na região occipital e alteração da coloração da pele na região abdominal. A história perinatal não pode ser questionada, pois a criança havia sido abandonada pela mãe biológica logo após o nascimento. Exame físico geral apresentava baixa estatura e desnutrição, microstomia, lábios finos, alopecia predominante em região occipital, temporal e frontal, redução dos cílios. No abdome, dorso e região lombar havia placas esclerodermiformes (Figura 1).
Foi realizada biópsia cutânea da região abdominal e o exame histopatológico evidenciou epiderme atrófica com hialinização da derme e da gordura subcutânea, redução das estruturas anexiais.
Os exames bioquímicos revelaram aumento de triglicerídeos e do colesterol total 286mg/dl e 230 mg/dl, respectivamente. Realizou ecocardiograma, radiografias das mãos e punhos e de tórax os quais não detectaram nenhuma anormalidade.
Aos 2 anos e 6 meses de idade, paciente evoluiu com alopecia universal tornando evidentes as veias proeminentes do couro cabeludo. A presença das alterações clínicas e dos achados laboratoriais permitiu o diagnóstico de Progeria e, desde então, encontra-se em acompanhamento multidisciplinar (Figura 2, 3, 4).
DISCUSSÃO
A Progeria ou Síndrome de Hutchinson-Gilford é uma doença autossômica dominante. Foi descrita, pela primeira vez, em 1886, por Hutchinson, e ratificada por Gilford, em 1904. Possui ocorrência esporádica, com incidência de 1 a cada 8 milhões de nascidos vivos, tendo aproximadamente 150 casos descritos na literatura, predomina no sexo masculino com relação de 1,5:1 e maior suscetibilidade dos caucasianos em 97% dos casos. 1, 2, 3, 4
Caracteriza-se por envelhecimento precoce, apresentando ritmo superior a sete vezes em relação à normalidade, causando alterações em vários órgãos e sistemas, como: a pele, tecido celular subcutâneo, pelos, sistema cardiovascular e esquelético. 1, 4, 5
A base genética foi descoberta em 2003, com achados de mutação no gene LMNA, o qual codifica a Lâmina A, gerando uma produção de uma proteína aberrante chamada progerina, classificando esta doença no grupo das laminopatias. A progerina está presente, em alta concentração, nas células desses pacientes, tanto que promove uma distorção na membrana nuclear, no que altera a função da cromatina e, assim, declina a expectativa de vida. 1, 3, 4, 5, 6
Os pacientes portadores desta doença são normais ao nascimento, manifestando as primeiras alterações, no final do primeiro ano de vida, quando o ganho ponderal e o crescimento reduzem, assim como a pele torna-se esclerodermiforme e surgem os primeiros sinais de alopecia. 1, 2, 4, 7
As manifestações clínicas são divididas em critérios maiores e sinais usualmente presentes. Estes são: face de pássaro (surgimento em torno de 6 meses a 1 ano de idade), alopecia, veias proeminentes no couro cabeludo, olhos grandes, micrognatia, dentição anormal e lenta, tórax em pera, clavículas curtas, pernas arqueadas (coxa valga), membros superiores curtos e articulações proeminentes, baixa estatura e peso com idade óssea normal, maturação sexual incompleta, redução do tecido adiposo e desenvolvimento psicomotor adequado com inteligência normal. 1,2, 4, 6, 8
O diagnóstico é essencialmente clínico, com surgimento dos critérios maiores no primeiro e segundo ano de vida. Como diagnóstico diferencial, enumeramos outras síndromes, como: a acrogeria; a pangeria e a síndrome Bloom, todas caracterizadas por envelhecimento precoce. 1, 2, 9
O prognóstico é reservado, com expectativa de vida em torno de 13 anos. O principal fator de mortalidade são as doenças cardiovasculares (75%) como o infarto agudo do miocárdio. Apesar dos avanços da cirurgia cardiovascular, a baixa taxa de sobrevida mantém-se em face de sua alta capacidade em refazer as placas ateromatosas. 2, 5, 8
Até o momento, não existe terapêutica específica e ela é dirigida somente às complicações. Os recentes avanços da biologia molecular com o reconhecimento das alterações genéticas podem melhorar o conhecimento do envelhecimento na espécie humana.
Como as primeiras manifestações são dermatológicas, 35% dos pais procuram o dermatologista, este deve estar atento para realizar precocemente o diagnóstico, como ocorreu no caso apresentado acima.

Aprovado pelo Conselho Editorial e aceito para publicação em 20.06.2010.
Conflito de interesse: Nenhum
Suporte financeiro: Nenhum
-
1Rastogi R, Mohan SMC. Progeria syndrome: A case report. Indian J Orthop. 2008;42:97-9.
-
2Pardo RAV, Castillo ST. Progeria. Rev Chil Pediatr. 2002;73:5-8.
-
3Delbarre E, Tramier M, Coppey-Moisan M, Gaillard C, Courvalin JC, Buendia B. The truncated prelamin A in Hutchinson-Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum Mol Genet. 2006;15:1113-22.
-
4Mazereeuw-Hautier J, Wilson LC, Mohammed S, Smallwood D, Shackleton S, Atherton DJ, et al. Hutchinson-Gilford progeria syndrome: clinical findings in three patients carrying the G608G mutation in LMNA and review of the literature. Br J Dermatol. 2007;156:1308-14.
-
5Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, et al. Disease Progression in Hutchinson-Gilford Progeria Syndrome: Impact on Growth and Development. Pediatrics. 2007;120;824-33.
-
6Kieran MW, Gordon L, Kleinman. New Approaches to Progeria. Pediatrics. 2007;120; 834-41.
-
7Giuar PJ, Kaye CI, McCourt JW. Progressive Early Dermatologic Changes in Hutchinson-Gilford Progeria Syndrome. Pediatr Dermatol. 1991;8:199-206.
-
8Ceballos LE, Pérez DME, Núñez AR. Progeria. Presentación de 1 caso. Rev Cuba Ortop Traumatol.1999;13:129-31.
-
9Salomão PR, Nogueira GC, Café MEM. Você conhece esta síndrome? Acrogeria. An Bras Dermatol. 2005;80:192-4.
Datas de Publicação
-
Publicação nesta coleção
21 Mar 2011 -
Data do Fascículo
Fev 2011
Histórico
-
Recebido
20 Jun 2010