
Resumos
A Síndrome de Bloch-Sulzberger (Incontinência Pigmentar) é uma genodermatose rara, que afeta, principalmente, o sexo feminino, pois costuma ser letal em pacientes do sexo masculino intraútero. Caracteriza-se, principalmente, pelas manifestações dermatológicas, podendo também apresentar anomalias dentárias, oftalmológicas e neurológicas. As lesões cutâneas apresentam 4 fases distintas: vesiculosa, verrucosa, pigmentar e atrófica; que podem seguir uma sequência irregular, havendo até sobreposição das mesmas
Dermatopatias genéticas; Incontinência pigmentar; Transtornos da pigmentação
Bloch-Sulzberger syndrome (incontinentia pigmenti) is a rare genodermatosis that affects predominantly females, since it is generally lethal to male fetuses in utero. It is characterized principally by skin lesions, but may also involve dental, ophthalmological and neurological abnormalities. The skin lesions are present in four different phases: vesicular, verrucous, hyperpigmented and atrophic/hypopigmented. Their sequence is irregular and overlapping of stages is common
Incontinentia pigmenti; Pigmentation disorders; Skin diseases, genetic
SÍNDROME EM QUESTÃO
Você conhece esta síndrome?* * Trabalho realizado no Hospital Municipal Jesus (HMJ) - Rio de Janeiro (RJ), Brasil.
Isabella Brasil SucciI; Fernando Colonna RosmanII; Elisa Fontenelle de OliveiraIII
IMédica dermatologista - Preceptora da pós-graduação em Dermatologia do Instituto de Dermatologia Prof. Azulay - Santa Casa da Misericórdia do Rio de Janeiro (IDPA-SCMRJ) - Rio de Janeiro (RJ), Brasil
IIPatologista do Hospital Municipal Jesus (HMJ) - Rio de Janeiro (RJ), Brasil
IIIDermatologista do Hospital Municipal Jesus (HMJ) - Responsável pelo ambulatório de dermatologia pediátrica do Instituto de Dermatologia Prof. Rubem David Azulay - Santa Casa da Casa da Misericórdia do Rio de Janeiro (IDPA - SCMRJ) - Professora Substituta no Hospital Universitário Pedro Ernesto - Universidade do Estado do Rio de Janeiro (HUPE - UERJ) - Rio de Janeiro (RJ), Brasil
Endereço para correspondência Endereço para correspondência: Isabella Brasil Succi Av. Visconde de Pirajá, 82/805 Ipanema Rio de Janeiro - CEP 22410-000 Tel:/Fax: (21) 25210191 (21) 25133370 Email: ibsucci@unisys.com.br
RESUMO
A Síndrome de Bloch-Sulzberger (Incontinência Pigmentar) é uma genodermatose rara, que afeta, principalmente, o sexo feminino, pois costuma ser letal em pacientes do sexo masculino intraútero. Caracteriza-se, principalmente, pelas manifestações dermatológicas, podendo também apresentar anomalias dentárias, oftalmológicas e neurológicas. As lesões cutâneas apresentam 4 fases distintas: vesiculosa, verrucosa, pigmentar e atrófica; que podem seguir uma sequência irregular, havendo até sobreposição das mesmas.
Palavras-chave: Dermatopatias genéticas; Incontinência pigmentar; Transtornos da pigmentação
RELATO DO CASO
Paciente lactente, 2 meses, apresentou ao nascimento vesículas, pústulas e bolhas disseminadas, inicialmente nos membros inferiores, com posterior disseminação para abdome e membros superiores. Recebeu o diagnóstico de impetigo e prescrição de cefalexina, evoluiu com aumento do número das lesões, sendo então encaminhada para o ambulatório de dermatopediatria. Ao exame dermatológico, notavam-se lesões verrucosas lineares, principalmente, na região flexora dos membros (Figuras 1A e 1B); apresentava também alopecia no vértex (Figura 2A).


A mãe apresentava áreas de hiperpigmentação na virilha (Figura 2B) e referia uma história de rash na infância.
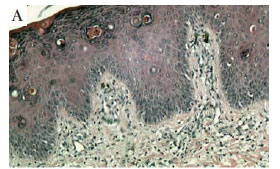
O hemograma evidenciou eosinofilia de 11%, a biópsia de uma lesão verrucosa na coxa direita mostrou espongiose, rico infiltrado eosinofílico e melanófagos (Figuras 3A e 3B).
DISCUSSÃO
A Síndrome de Bloch-Sulzberger (Incontinência Pigmentar) é uma desordem ectodérmica, dominante, ligada ao X. Afeta a pele, os olhos, os dentes e pode estar associada a déficits neurológicos.1,2 Incidência aproximada de 1:50000 nascidos, sendo, tipicamente, letal intraútero no sexo masculino. História familial é encontrada em cerca de 50% dos casos.2
O nome incontinência pigmentar (IP) descreve uma característica histológica, não específica, onde há, na derme superficial, incontinência de melanina dos melanócitos da camada basal.3
O Fator Nuclear kB (NF-kB) regula a expressão de múltiplos genes, incluindo citocinas e moléculas de adesão. A ativação do NF-kB também confere proteção contra a apoptose induzida pelo Fator de Necrose Tumoral (TNF). O gen NEMO é essencial no processo de ativação do NF-kB.4,5 Recentemente, a causa da IP foi atribuída a uma mutação que inativa o gen NEMO, localizado no cromossomo X (q28).2
A apresentação cutânea da IP regride espontaneamente pela eliminação seletiva das células que apresentam o cromossomo X mutante, havendo substituição gradual por células normais.2 Geralmente, os achados dermatológicos são o primeiro sinal observado, estando presente em, praticamente, todos os pacientes.4
Em 90% dos casos, nas duas primeiras semanas de vida, as lesões cutâneas surgem e são caracterizadas por quatro fases (vesiculosa, verrucosa, pigmentar e atrófica) que podem seguir uma sequência irregular, com duração variável, havendo até sobreposição das mesmas.1,6
Na primeira, há vesículas ou pústulas geralmente precedidas por eritema, que podem ocorrer em qualquer local do corpo, poupando a face. Desenvolvem-se nas primeiras semanas de vida, desaparecendo por volta do 4ºmês. Há distribuição linear nos membros e circulares no tronco. Encontra-se infiltração de eosinófilos nas vesículas epidérmicas e leucocitose no sangue periférico, com eosinofilia.1,3 Costuma ser confundido com lesões herpéticas ou infecções bacterianas.4
Em 70% dos casos, a fase vesiculosa é seguida pela verrucosa, aparecendo, geralmente, na parte distal dos membros, assim que as bolhas começam a cicatrizar, tornando-se secas e hiperceratóticas. Raramente, afetam tronco e face, mas podem ocorrer no couro cabeludo.3 Esta fase dura algumas semanas, com clareamento das lesões aos 6 meses de idade em 80% dos pacientes.4
A fase seguinte acomete 98% dos pacientes, havendo hiperpigmentação que costuma desaparecer no final da segunda década. As lesões são circulares e lineares, acastanhadas, seguindo as linhas de Blaschko.4 São mais aparentes no tronco do que nos membros. As nádegas estão frequentemente envolvidas. Invariavelmente, axilas e virilhas são afetadas.3 As áreas hiperpigmentadas, geralmente, não correspondem às áreas previamente acometidas com vesículas ou inflamação, não representando um processo de hiperpigmentação pós-inflamatória.4
A quarta fase se caracteriza por lesões lineares atróficas, hipocrômicas, com ausência de pêlos. Acomete as extremidades em 77% dos casos, ocorrendo em adolescentes e adultos. Estas alterações costumam ser permanentes, sendo o único sinal de envolvimento cutâneo da doença em adultos.4
A manifestação mais comum nos cabelos é a alopecia no vértex, ocorrendo em 38% dos pacientes. Nas unhas podem surgir depressões puntiformes ou sulcos, que podem desaparecer com a idade. Também observamos tumores periungueais ou subungueais. As unhas das mãos são mais afetadas do que as dos pés.4
Os dentes estão acometidos em 80% dos casos, sendo a manifestação não cutânea mais comum da IP. Estas alterações são permanentes, ao contrário das lesões da pele. Ambos os dentes decíduos e permanentes são afetados, sendo comum hipo ou anodontia.4 Dentes cônicos ocorrem em 30% dos casos.
As alterações oftalmológicas estão entre as manifestações mais graves da doença. O envolvimento assimétrico é comum. Pode ocorrer estrabismo, nistagmo, catarata, atrofia do nervo óptico. As lesões da retina são resultantes de eventos vaso-oclusivos. Frequentemente, estas manifestações oftalmológicas estão associadas a déficits neurológicos, que incluem convulsões, paralisia espástica, retardo motor e microcéfalo.4 A incidência de anormalidades neurológicas é menor em casos de IP familiar.3
Exames de imagem, como: tomografia computadorizada, ressonância nuclear magnética e angiografia são úteis para avaliar o envolvimento oftalmológico e neurológico; assim como eletroencefalograma, em pacientes com convulsão.4
Para o diagnóstico, há critérios propostos, quando não há evidência de IP em parente feminino de1º grau, sendo critérios maiores:
- Eritema e vesículas com eosinofilia.
- Hiperpigmentação típica.
- Lesões atróficas. Os critérios menores incluem:
- Anormalidades dentárias.
- Alopecia.
- Anormalidades ungueais.
- Alterações na retina.
Havendo evidência de IP em parente feminina de 1ºgrau os critérios incluem:
- História sugestiva de rash típico.
- Hiperpigmentação, lesões atróficas.
- Alopecia no vértex.
- Anormalidades dentárias.
- Alterações na retina.
- Múltiplos abortos de fetos masculinos.4
O tratamento das lesões cutâneas é sintomático com emoliente e antibiótico, em casos de infecção secundária.6,7 As alterações vasculares da retina são progressivas durante os primeiros meses de vida, estando recomendado exames mensais neste período. Os pais devem ser advertidos sobre uma possível ausência de dentes ou dentição tardia.3

Aprovado pelo Conselho Editorial e aceito para publicação em 04.10.2010.
Conflito de interesse: Nenhum
Suporte financeiro: Nenhum
- 1. Paller AS, Mancini AJ. Disorders of Both Hypopigmentation and Hyperpigmentation. In: Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology. 3rd. ed. Philadelphia: Elsevier Saunders; 2006.p.280-3.
- 2. Nogueira A, Lisboa C, Eloy C, Mota A, Azevedo F. Vesicular rash in a newborn. Indian J Dermatol Venereol Leprol. 2009;75:330.
- 3. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger Syndrome). J Med Genet. 1993;30:53-9.
- 4. Berlin A, Paller A, Chan L. Incontinentia pigmenti: A review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002;47:169-87.
- 5. Pereira MAC, Mesquita LAF, Budel AR, Cabral CSP, Feltrim AS. Incontinencia pigmentar ligada ao X ou síndrome de Bloch-Sulzberger: relato de um caso. An Bras Dermatol. 2010;85:372-5.
- 6. Bleehen SS, Anstey AV. Incontinentia pigmenti. In: Burns T, Breathnach S, Cox N, Griffiths C, editors. Rook's Textbook of Dermatology. 7th ed. London: Blackwell Publishing; 2004. p.39.20-2.
- 7. Sampaio SAP, Rivitti EA. Alterações Hereditárias Mesenquimais e Pigmentares e Malformações. In: Sampaio SAP, Castro RM, Rivitti EA. Dermatologia. 2. ed. São Paulo: Artes Médicas; 2000.p.807-8.
Datas de Publicação
-
Publicação nesta coleção
22 Jun 2011 -
Data do Fascículo
Jun 2011
Histórico
-
Recebido
04 Out 2010



