Abstracts
Stiff skin syndrome is a rare scleroderma-like disorder of unknown etiology characterized by stone-hard indurations of skin, mild hypertrichosis and limited joint mobility. No effective treatment has yet been found. Exercises and rehabilitative therapy are important in maintaining the patient's quality of life. The authors present a case of a two-year-old boy with progressive skin hardening since he was eightmonth old and secondary restricted joint mobility, diagnosed as Stiff skin syndrome
Contracture; Fascia; Mucopolysaccharidoses; Rare diseases; Scleroderma; systemic
Síndrome stiff skin é doença rara, esclerodermiforme, de etiologia desconhecida, caracterizada por endurecimento pétreo da pele, hipertricose leve e limitação da mobilidade articular. Não há tratamento efetivo até o momento. Exercícios e reabilitação são importantes para manter a qualidade de vida do paciente. Os autores apresentam caso de um menino de dois anos de idade com endurecimento cutâneo progressivo desde os oito meses de idade e restrição secundária da mobilidade articular, diagnosticado como Síndrome stiff skin
Contratura; Doenças Raras; Escleroderma sistêmico; Fascia; Mucopolissacaridoses
CASE REPORT
Stiff skin syndrome - case report*
Adriana Gutstein da Fonseca AmorimI; Marcia Kalil AidéII; Sandra Maria Barbosa DurãesIII; Mayra Carrijo RochaelIV
IThird year Specialist Trainee in Dermatology at the Universidade Federal Fluminense (UFF) - Niterói (RJ), Brasil
IIMedical Doctor of the Hospital de Força Aérea do Galeão (HFAG) - Galeão (RJ), Brasil
IIIPhD - Assistant Teacher in Dermatology at the Universidade Federal Fluminense (UFF) - Niterói (RJ), Brasil
IVPhD in Anatomopathology - Associate Teacher of the Universidade Federal Fluminense (UFF) - Niterói (RJ), Brasil
Mailing address
ABSTRACT
Stiff skin syndrome is a rare scleroderma-like disorder of unknown etiology characterized by stone-hard indurations of skin, mild hypertrichosis and limited joint mobility. No effective treatment has yet been found. Exercises and rehabilitative therapy are important in maintaining the patient's quality of life. The authors present a case of a two-year-old boy with progressive skin hardening since he was eightmonth old and secondary restricted joint mobility, diagnosed as Stiff skin syndrome.
Keywords: Contracture; Fascia; Mucopolysaccharidoses; Rare diseases; Scleroderma, systemic
INTRODUCTION
Stiff skin Syndrome (SSS) is a rare disease that is present at birth or early infancy, characterized by stone-hard skin, especially in areas with abundant fascia like buttocks and thighs, limitation of joint mobility secondary to the skin thickening and mild hypertrichosis. 1 The cutaneous involvement is not associated to visceral, muscular, immunological or vascular changes. 2 We report a case of child with clinical and histopathological characteristics of SSS.
CASE REPORT
Two year old male black infant, original from Niterói, Rio de Janeiro. He was sent to the Dermatology service for the evaluation of asymptomatic skin hardening on the upper abdomen and thighs, noticed by the mother since the age of eight months. The patient was born at term after an uneventful pregnancy. He was the only child of healthy, non-consanguineous parents. There was no history of previous traumas to the affected areas, frequent use of any medication, infectious diseases or convulsions. The patient's cognitive development was appropriate for his age but his height and weight were below the expected. There was no report of Raynaud's phenomenon and no familiar case of diabetes, scleroderma or similar cutaneous findings.
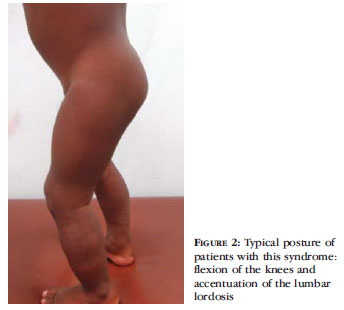
At physical examination the patient had a small, hard, non folding plaque on the skin of the upper abdomen and a rock-hard indurated area around the thighs, close to the knees, as well as slight thickening of the skin on the buttocks, without hypertrichosis. There was a noticeable shortening of the left leg in relation to the right, "genu varum" deformity and joint stiffness with restriction of the amplitude of the movement of the legs and hips (Figure 1). On standing position the child would show fixed flexion of the knees and accentuation of the lumbar lordosis (Figure 2). There was no alteration at the neurological examination. Routine blood tests including complete haemogram, erythrocyte sedimentation rate (ESR), glucose, urea, creatinine, lipoprotein panel, C-reactive protein, SGPT, SGOT, bilirrubines, alkaline phosphatase, glutamyl transferase, albumin, protein electrophoresis, thyroid tests, antistreptolysin O, stool and urine exams were all within normal range. Dosages of mucopolysaccharides (on blood and urine), muscle enzymes, anti-double stranded DNA antibody, anti-centromere, anti-Scl-70, anti-Ro, anti-La and chest x-ray were normal. Due to the age of the patient computerized tomography and electromyography were not performed. Ultrasound and Doppler of the abdomen and pelvis were normal, except for the thickening of the soft tissues, up to the aponeurosis, which was also present on the ultrasound of soft tissues on the lower limbs.
Histopathological examination of biopsied samples from lesions on the abdomen and right thigh stained by haematoxylin and eosin (HE) showed normal epidermis, thickening of the collagen on the dermis with normal appendages and vessels and no inflammatory infiltrate (Figure 3). Staining by Alcian Blue at ph 2.5 revealed moderate deposits of mucopolyssacharides on the dermis between the collagen bundles, making it impossible to visualize the fascia. The combined histological and clinical findings were suggestive of Stiff Skin Syndrome (Figure 4).
The therapeutic plan for the patient included physiotherapy and physical exercise. Initially the mother did not follow the recommendations and after eight months of the diagnosis the deformity of the patient's legs worsened. Treatment based on physiotherapy was started once a week and physical exercise like soccer was encouraged, which led to an improvement in walking. After one year expanding of the lesions around the knees and involvement of almost the entire abdomen, with the aspect of "cobblestone" were noticed at physical examination (Figure 5). The mother was instructed to increase the physiotherapy sessions and come back in three months, but she discontinued the treatment despite phone calls and medical recommendation.
DISCUSSION
SSS is an uncommon connective tissue disease similar to scleroderma, described in 1971 by Esterly and Mc Kusik. Visceral and muscular involvement, vascular hyperactivity and immune abnormalities are absent. The clinical presentation is heterogeneous and there are no pathognomonic histological or laboratorial findings, thus being a diagnostic of exclusion. The age of the start of the symptoms varies from birth to childhood and there is no predilection for sex or race. The clinical criteria that support the diagnosis of the syndrome are: 1-hereditary condition, 2-stone-hard thickening of the skin more prominent in areas with abundant fascia like buttocks and thighs, 3-limitation of joint mobility secondary to the thickening of the overlying skin, 4-absence of urinary mucopolysaccharides, 5- mild and variable hypertrichosis. The patients might have a delayed growth with normal neuropsychomotor development, typical "tiptoe'' posture, accentuation of the lumbar lordosis, cutaneous lesions with the ''cobblestone'' pattern and restrictive pulmonary abnormalities secondary to the stiffening of the thoracic skin. 1, 3, 4, 5
The pathogenesis of SSS remains unknown. The two main pathogenic hypotheses are: a primary abnormality of the fascia with increased production of collagen VI or a congenital abnormality of the fibroblasts leading to a non-inflammatory dermal fibrosis due to a defective synthesis of mucopolysaccharides. 2,3,6-8 Some authors have detected high levels of proinflammatory cytokines (tumor necrosis factor-alpha, Inteleukin-6 and transforming growth factor-β2) suggesting that the fibrosis might be related to the inflammatory process. However, systemic corticosteroids and immunosuppressant drugs do not modify the progress of the syndrome. 2, 4, 9
There are familial and consanguineous reports in 30% of the cases, which suggests the possibility of genetic transmission. 1, 2, 6 Loeys and collaborators presented the results of a genetic study with evidence that a mutation of the locus 15q21.1 of the FBN1 gen that regulates the production of the profibrotic cytokine transforming growth factor-β2 (TGF- β2) causes the SSS.9
Two variants of SSS have been described: The Parana Syndrome, which has a worse clinical progress, with diffuse cutaneous involvement, restrictive pulmonary abnormalities, slow growth and consanguinity, and the congenital fascial dystrophy, that involves isolated areas of abundant fascia. 2, 4, 7, 10-13
The histology might show various levels of fibrosis on the dermis and subcutaneous tissue, with the presence of mucopolyssacharides amongst the collagen fibers at earlier stages, absence of inflammatory infiltrate and preservation of the blood vessels and cutaneous appendages. The fascia might or might not be thickened.4
The patient fulfilled the clinical criteria for SSS although he did not have hypertrichosis, which is a variable finding, developing over hardened skin areas. He had the abdominal lesions with the characteristic ''cobblestone'' pattern as well as an accentuation of the lumbar lordosis and flexion of the knees, typical of SSS. There was no report of consanguinity or familial cases. The laboratory and image tests did not show any signs of immunological, inflammatory, muscular or visceral alteration. The histopathology probably represents the initial stage of the syndrome with the presence of mucin on the dermis, since at advanced stages there is sclerosis of the deep dermis.
The main differential diagnosis of SSS are the systemic scleroderma, which is rare in childhood and is associated with cutaneous lesions on the face and hands, Raynaud's phenomenon, periungual telangectasias and visceral and immunological abnormalities, which were absent in this patient. 4, 14 The localized scleroderma, also known as morphea, presents with mostly cutaneous involvement and only rarely involves the joints, causing contractures and retraction of the limbs. 15 The histology differs from SSS, with lymphocytic infiltrate on the dermal-epidermal junction and abnormally sclerotic collagen on the reticular dermis, and it responds favorably to treatment with anti inflammatory medication. Neonatal scleredema is more related to prematurity, and the histology shows lesions to the adipocytes, which were absent on the actual patient. 16 Scleredema usually starts abruptly and results from infiltration of mucin into the dermis, causing the clinical aspect of non-pitting edema and more uniform cutaneous hardening, not as well defined as in SSS, predominantly on the upper trunk, neck and face (unusual locations for the syndrome), and develops mostly in adults. Sclerodermatomyositis has characteristics of both scleroderma and dermatomyositis, with the elevation of muscular enzymes, auto antibodies and vascular hyper reactivity. Scleromyxedema, or lichen myxedematosus, shows progressive thickening of the face, neck, ears, upper limbs and upper trunk, as well as a papulous eruption. 17 The mucopolysaccharidosis can present with nodules and plaques on the extremities or upper trunk, with the ''orange peel ' 'aspect and the presence of mucopolysaccharides on the urine and blood.5, 8
The treatment described on the literature, based on motor physiotherapy, was proposed during the ambulatory follow-up and provided an improvement on the quality of life of the patient, but the cutaneous thickening progressed slowly, as seen on the literature. There are many reports of treatments aiming at slowing down the progression of the disease, like immunosuppressant drugs, corticotherapy, psoralens and penicillamine, however the patients have not shown any clinical improvement. 5 The authors have found 43 cases of SSS on the world literature, five of which reported in Brazil. 
REFERENCES
-
1Esterly NB, McKusick VA. Stiff skin syndrome. Pediatrics. 1971;47:360-9.
-
2Jablonska S, Schubert H, Kikuchi I. Congenital fascial dystrophy: stiff skin syndrome- a human counterpart of the tight-skin mouse. J Am Acad Dermatol. 1989;21:943-50.
-
3Jablonska S, Groniowski J, Krieg T, Nerlich A, Peltonen L, Oikarinen A, et al. Congenital fascial dystrophy-a non inflammatory disease of fascia: the stiff skin syndrome. Pediatr Dermatol. 1984;2:87-97.
-
4Richard MA, Grob JJ, Philip N, Rey J, Chamson A, Mege JL, et al. Physiopathogenic investigations in a case of familial stiff-skin syndrome. Dermatology. 1998;197:127-31.
-
5Liu T, Mc Calmont TH, Frieden IJ, Williams ML, Connolly MK, Gilliam AE. The stiff skin syndrome:case series, differential diagnosis of the stiff skin phenotype, and review of the literature. Arch Dermatol. 2008;144:1351-9.
-
6Kikuchi I, Inoue S, Hamada K, Ando H. Stiff skin syndrome. Pediatr Dermatol. 1985;3:48-53.
-
7Jablonska S, Blaszczyk M. Scleroderma-like indurations involving fascias: an abortive form of congenital fascial dystrophy (Stiff skin syndrome). Pediatr Dermatol. 2000;17:105-10.
-
8Morrell DS, Challgren E, Nijhawan A, Olson J, Laumann A, Medenica M, et al. Two cases for diagnosis: asymmetric childhood scleredema or Stiff skin syndrome? Pediatr Dermatol. 2003;20:350-5.
-
9Loeys BL, Gerber EE, Riegert-Johnson D, Iqbal S, Whiteman P, McConnell V, et al. Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci Transl Med. 2010;2:23ra20.
-
10Cat I, Magdalena NI, Marinoni LP, Wong MP, Freitas OT, Malfi A, et al. Parana hardskin syndrome: study of seven families. Lancet. 1974;303:215-16.
-
11Jablonska S, Blaszczyk M. Stiff skin syndrome is highly heterogeneous, and congenital fascial dystrophy is its distinct subset. Pediatr Dermatol. 2004;21:508-10.
-
12Guiducci S, Distler JH, Milia AF, Miniati I, Rogai V, Manetti M, et al. Stiff skin syndrome: evidence for an inflammation-independent fibrosis? Rheumatology. 2009;48:849-52.
-
13Fidzianska A, Jablonska S. Congenital fascial dystrophy: abnormal composition of the fascia. J Am Acad Dermatol. 2000;43:797-802.
-
14Azevedo VF, Serafini SZ, Werner B, Muller CS, Franchini CFM, Morais RLSL. Stiff skin syndrome versus scleroderma: a report of two cases. Clin Rheumatol. 2009;28:1107-11.
-
15Zancanaro PCQ, Isaac AR, Garcia LT, Costa IMC. Esclerodermia localizada na criança: aspectos clínicos, diagnósticos e terapêuticos. An Bras Dermatol. 2009;84:161-72.
-
16Heilbron B, Saxe N. Scleredema in an infant. Arch Dermatol. 1986;122:1417-19.
-
17Bopp C. Mucinoses cutâneas. An Bras Dermatol. 1968;43:7-24.
Publication Dates
-
Publication in this collection
08 Nov 2011 -
Date of issue
Aug 2011
History
-
Received
26 Aug 2010 -
Accepted
01 Nov 2010