Resumos
Síndrome stiff skin é doença rara, esclerodermiforme, de etiologia desconhecida, caracterizada por endurecimento pétreo da pele, hipertricose leve e limitação da mobilidade articular. Não há tratamento efetivo até o momento. Exercícios e reabilitação são importantes para manter a qualidade de vida do paciente. Os autores apresentam caso de um menino de dois anos de idade com endurecimento cutâneo progressivo desde os oito meses de idade e restrição secundária da mobilidade articular, diagnosticado como Síndrome stiff skin
Contratura; Doenças Raras; Escleroderma sistêmico; Fascia; Mucopolissacaridoses
Stiff skin syndrome is a rare scleroderma-like disorder of unknown etiology characterized by stone-hard indurations of skin, mild hypertrichosis and limited joint mobility. No effective treatment has yet been found. Exercises and rehabilitative therapy are important in maintaining the patient's quality of life. The authors present a case of a two-year-old boy with progressive skin hardening since he was eightmonth old and secondary restricted joint mobility, diagnosed as Stiff skin syndrome
Contracture; Fascia; Mucopolysaccharidoses; Rare diseases; Scleroderma; systemic
CASO CLÍNICO
Síndrome stiff skin : relato de caso* * Trabalho realizado no Hospital Universitário Antonio Pedro - Universidade Federal Fluminense (HUAP - UFF) - Niterói (RJ), Brasil.
Adriana Gutstein da Fonseca AmorimI; Marcia Kalil AidéII; Sandra Maria Barbosa DurãesIII; Mayra Carrijo RochaelIV
IEspecializanda do terceiro ano de Dermatologia da Universidade Federal Fluminense (UFF) - Niterói (RJ), Brasil
IIMédica do Hospital de Força Aérea do Galeão (HFAG) - Galeão (RJ), Brasil
IIIDoutorado - Professora-adjunta de Dermatologia da Universidade Federal Fluminense (UFF) - Niterói (RJ), Brasil
IVDoutora em Anatomia Patológica - Professora-associada da Universidade Federal Fluminense (UFF) - Niterói (RJ), Brasil
Endereço para correspondência Endereço para correspondência: Adriana Gutstein da Fonseca Amorim E-mail: adrianadermatologista@gmail.com
RESUMO
Síndrome stiff skin é doença rara, esclerodermiforme, de etiologia desconhecida, caracterizada por endurecimento pétreo da pele, hipertricose leve e limitação da mobilidade articular. Não há tratamento efetivo até o momento. Exercícios e reabilitação são importantes para manter a qualidade de vida do paciente. Os autores apresentam caso de um menino de dois anos de idade com endurecimento cutâneo progressivo desde os oito meses de idade e restrição secundária da mobilidade articular, diagnosticado como Síndrome stiff skin.
Palavras-chave: Contratura; Doenças Raras; Escleroderma sistêmico; Fascia; Mucopolissacaridoses
INTRODUÇÃO
Síndrome stiff skin (SSS) é doença rara que se apresenta ao nascimento ou infância precoce, caracterizada por pele pétrea, especialmente em áreas com abundante fáscia, como nádegas e coxas, limitação da mobilidade articular secundária ao espessamento cutâneo e hipertricose leve.1 O envolvimento cutâneo não se associa a alterações viscerais, musculares, imunológicas ou vasculares.2 Relata-se caso de uma criança com características clínicas e histopatológicas de SSS.
RELATO DE CASO
Criança negra de dois anos de idade, sexo masculino, procedente de Niterói, Rio de Janeiro, encaminhada ao Ambulatório de Dermatologia para avaliar endurecimento assintomático da pele do abdome superior e coxas, percebido pela mãe desde os 8 meses de idade. Nascido a termo, após gestação sem intercorrências. Filho único de pais saudáveis e sem consanguinidade. História patológica pregressa negativa para traumas na área afetada, medicamentos de uso frequente, doenças infecciosas ou convulsões. O paciente apresentava desenvolvimento cognitivo apropriado para a idade, porém, baixo peso e baixa estatura. Não havia relato de fenômeno de Raynaud ou outros sintomas na revisão de sistemas e nenhum caso familiar de diabetes, esclerodermia ou quadros cutâneos semelhantes.
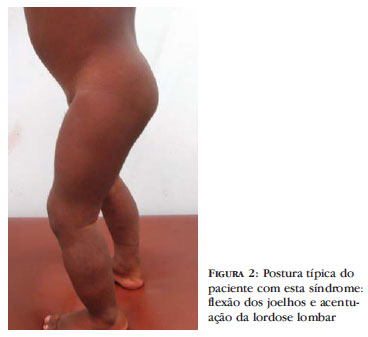
No exame físico, a pele do abdome superior apresentava pequena placa de endurecimento não pregueável e área endurecida, pétrea, circundando as coxas, próxima aos joelhos, além de discreto espessamento da pele das nádegas, sem hipertricose. Havia visível encurtamento da perna esquerda em relação à direita, deformidade em "genu varum" e rigidez articular com restrição de amplitude de movimentos das pernas e quadril (Figura 1). Na posição ortostática, adotava postura de flexão fixa de joelhos e acentuação da lordose lombar (Figura 2). O exame neurológico não apresentava alterações. Os exames laboratoriais de rotina, incluindo hemograma completo, VHS, glicose, ureia, creatinina, lipidograma, proteína C reativa, TGO, TGP, bilirrubinas, fosfatase alcalina, gama GT, albumina, eletroforese de proteínas, hormônio tireoidiano, ASO (antiestreptolisina) e exame de fezes e urina estavam dentro dos padrões normais. A dosagem de mucopolissacarídeos (no sangue e na urina), enzimas musculares, anti-DNA dupla hélice, anticentrômero, anti-Scl 70, anti-Ro, anti-La e radiografia de tórax eram normais. Devido à idade do paciente, optou-se por não ser realizadas tomografia computadorizada e eletroneuromiografia. Ultrassonografia e doppler de abdome e pelve foram normais, exceto pelo espessamento de partes moles até a aponeurose, também presente na ultrassonografia de partes moles de membros inferiores.
As biópsias das lesões do abdome e da coxa direita revelaram, aos cortes histológicos corados pela Hematoxilina e Eosina, epiderme normal, espessamento do colágeno da derme com preservação de apêndices, vasos normais e ausência de infiltrado inflamatório (Figura 3). A coloração Alcian Blue em pH 2,5 revelou depósito moderado de mucopolissacarídeos na derme entre os feixes de colágeno, não sendo possível visualizar a fáscia. O conjunto destes achados histopatológicos e clínicos foi sugestivo de Síndrome stiff skin (Figura 4).
A proposta terapêutica para o paciente incluiu fisioterapia motora e prática de exercícios físicos. Inicialmente, a mãe não seguiu as orientações, mas, após 8 meses do diagnóstico, observou uma piora da deformidade das pernas do paciente. Foi iniciado tratamento com fisioterapia motora uma vez por semana e estimulou-se a prática de esportes como futebol, observando-se, desde então, melhora na deambulação. Após um ano, ao exame físico, havia extensão das lesões ao redor dos joelhos e acometimento de quase todo o abdome, caracterizando aspecto em "pedra de calçamento" (Figura 5). A mãe foi orientada a aumentar as sessões de fisioterapia e retornar em três meses, mas descontinuou o acompanhamento, apesar dos contatos telefônicos e das orientações médicas.
DISCUSSÃO
SSS é uma doença incomum do tecido conectivo semelhante à esclerodermia, descrita em 1971 por Esterly e Mc Kusik. Envolvimento visceral, muscular, hiper-reatividade vascular e anormalidades imunológicas estão ausentes. A apresentação clínica é heterogênea e não há achados histopatológicos ou laboratoriais patognomônicos, sendo um diagnóstico de exclusão. A idade do início dos sintomas varia desde o nascimento até a infância, sem predileção por sexo ou raça. Os critérios clínicos que auxiliam o diagnóstico da síndrome são: 1-condição hereditária, 2-espessamento cutâneo pétreo mais proeminente em áreas de fáscia abundante como nádegas e coxas, 3-limitação da mobilidade articular secundária ao espessamento da pele sobrejacente, 4-ausência de mucopolissacarídeos urinários, 5- hipertricose leve e variável. Os pacientes podem apresentar retardo de crescimento com desenvolvimento neuropsicomotor normal, postura típica na ponta dos pés denominada "tiptoe", acentuação da lordose lombar, aparência das lesões cutâneas em "pedra de calçamento" e alterações restritivas pulmonares secundárias ao enrijecimento cutâneo do tórax. 1, 3, 4, 5
A patogênese da SSS permanece desconhecida. As duas principais hipóteses patogênicas são: uma anormalidade primária da fáscia, com superprodução de colágeno VI ou desordem congênita dos fibroblastos levando à fibrose dérmica não inflamatória pela síntese defeituosa de mucopolissacarídeos.2,3, 6-8 Alguns autores detectaram altos níveis de citocinas pró-inflamatórias (fator de necrose tumoral-alfa, interleucina-6 e fator transformador de crescimento-beta 2), sugerindo que a fibrose possa estar relacionada a processo inflamatório. Entretanto, os corticoides sistêmicos ou imunossupressores não modificam a evolução da síndrome. 2, 4, 9
Há casos familiares e de consanguinidade em 30% dos casos; por essa razão, vem sendo sugerida a possibilidade de transmissão genética.1, 2, 6 Loeys e colaboradores apresentaram resultados de pesquisa genética com evidências que indicam que a mutação no gene FBN1 do lócus 15q21.1, que regula a produção de citocina pró-fibrótica TGF-beta (fator transformador de crescimento beta), cause a SSS.9
Foram descritas duas variantes da SSS: a Síndrome Paraná, de pior evolução clínica, com acometimento cutâneo difuso, alterações pulmonares restritivas, retardo de crescimento e relatos de consangüinidade, e a distrofia fascial congênita, que acomete isoladamente áreas com abundante fáscia. 2, 4, 7, 10-13
A histopatologia pode apresentar graus variados de fibrose da derme e do tecido celular subcutâneo, com a presença de mucopolissacarídeos entre as fibras colágenas nos estágios iniciais, ausência de infiltrado inflamatório e preservação dos vasos sanguíneos e apêndices cutâneos. A fáscia pode ou não estar espessada.4
O paciente preencheu os critérios clínicos da SSS, mas não apresentava hipertricose, que é um achado variável e que pode ocorrer sobre as áreas de espessamento cutâneo. Possuía lesões abdominais caracterizando aspecto em "pedra de calçamento", assim como uma acentuação da lordose lombar e flexão de joelhos, típicas da SSS. Não se constatou consanguinidade e outros casos familiares. Em relação aos achados laboratoriais e de imagem, não foram detectadas alterações imunológicas, inflamatórias, musculares ou viscerais. A histopatologia corresponde provavelmente ao estágio inicial da síndrome pela presença de mucina na derme, pois nos estágios avançados há esclerose da derme profunda.
A SSS deve ser diferenciada principalmente da esclerodermia sistêmica, que é rara na infância, e está associada a alterações cutâneas na face e nas mãos, fenômeno de Raynaud, telangiectasias periungueais e alterações viscerais e imunológicas, as quais estavam ausentes no paciente. 4, 14 A esclerodermia localizada, também conhecida como morfeia, apresenta envolvimento predominantemente cutâneo, raramente podendo acometer articulações, ocasionando contraturas e retrações dos membros.15 Sua histologia é diferente da SSS, com infiltrado linfocítico na junção dermoepidérmica e colágeno anormalmente esclerótico na derme reticular e responde favoravelmente ao tratamento com medicamentos anti-inflamatórios. Escleredema neonatorum está mais relacionado à prematuridade e a histologia apresenta lesão de adipócitos, ausentes no paciente descrito.16 O escleredema costuma ter início abrupto e resulta de infiltração de mucina na derme, causando um quadro clínico de edema não depressível e endurecimento cutâneo mais uniforme, não tão bem definido como na SSS, predominando no tronco superior, pescoço e face (localizações incomuns da síndrome), ocorrendo principalmente em adultos. A esclerodermatomiosite tem características de esclerodermia e de dermatomiosite, com elevação de enzimas musculares, autoanticorpos e hiper-reatividade vascular. Escleromixedema, ou mixedema liquenoide, apresenta-se com espessamento progressivo da face, pescoço, pavilhões auriculares, membros superiores e tórax e erupção papulosa.17 As mucopolissacaridoses podem cursar com nódulos e placas nas extremidades ou tronco superior, de aspecto em "casca de laranja" e pela presença de mucopolissacarídeos urinários e sanguíneos.5, 8
O tratamento descrito na literatura com fisioterapia motora foi proposto durante o acompanhamento ambulatorial e proporcionou uma melhora na qualidade de vida do paciente, mas o espessamento cutâneo continuou progredindo lentamente, conforme a literatura. Há vários relatos de tentativas de frear a doença com imunossupressores, corticoterapia, psoralenos e penicilamina sem, contudo, ser observada a melhora dos pacientes. 5 Os autores encontraram 43 casos de SSS na literatura mundial, sendo cinco descritos no Brasil. 
Recebido em 26.8.2010.
Aprovado pelo Conselho Consultivo e aceito para publicação em 01.11.10.
Conflito de interesse: Nenhum
Suporte financeiro: Nenhum
-
1Esterly NB, McKusick VA. Stiff skin syndrome. Pediatrics. 1971;47:360-9.
-
2Jablonska S, Schubert H, Kikuchi I. Congenital fascial dystrophy: stiff skin syndrome- a human counterpart of the tight-skin mouse. J Am Acad Dermatol. 1989;21:943-50.
-
3Jablonska S, Groniowski J, Krieg T, Nerlich A, Peltonen L, Oikarinen A, et al. Congenital fascial dystrophy-a non inflammatory disease of fascia: the stiff skin syndrome. Pediatr Dermatol. 1984;2:87-97.
-
4Richard MA, Grob JJ, Philip N, Rey J, Chamson A, Mege JL, et al. Physiopathogenic investigations in a case of familial stiff-skin syndrome. Dermatology. 1998;197:127-31.
-
5Liu T, Mc Calmont TH, Frieden IJ, Williams ML, Connolly MK, Gilliam AE. The stiff skin syndrome:case series, differential diagnosis of the stiff skin phenotype, and review of the literature. Arch Dermatol. 2008;144:1351-9.
-
6Kikuchi I, Inoue S, Hamada K, Ando H. Stiff skin syndrome. Pediatr Dermatol. 1985;3:48-53.
-
7Jablonska S, Blaszczyk M. Scleroderma-like indurations involving fascias: an abortive form of congenital fascial dystrophy (Stiff skin syndrome). Pediatr Dermatol. 2000;17:105-10.
-
8Morrell DS, Challgren E, Nijhawan A, Olson J, Laumann A, Medenica M, et al. Two cases for diagnosis: asymmetric childhood scleredema or Stiff skin syndrome? Pediatr Dermatol. 2003;20:350-5.
-
9Loeys BL, Gerber EE, Riegert-Johnson D, Iqbal S, Whiteman P, McConnell V, et al. Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci Transl Med. 2010;2:23ra20.
-
10Cat I, Magdalena NI, Marinoni LP, Wong MP, Freitas OT, Malfi A, et al. Parana hardskin syndrome: study of seven families. Lancet. 1974;303:215-16.
-
11Jablonska S, Blaszczyk M. Stiff skin syndrome is highly heterogeneous, and congenital fascial dystrophy is its distinct subset. Pediatr Dermatol. 2004;21:508-10.
-
12Guiducci S, Distler JH, Milia AF, Miniati I, Rogai V, Manetti M, et al. Stiff skin syndrome: evidence for an inflammation-independent fibrosis? Rheumatology. 2009;48:849-52.
-
13Fidzianska A, Jablonska S. Congenital fascial dystrophy: abnormal composition of the fascia. J Am Acad Dermatol. 2000;43:797-802.
-
14Azevedo VF, Serafini SZ, Werner B, Muller CS, Franchini CFM, Morais RLSL. Stiff skin syndrome versus scleroderma: a report of two cases. Clin Rheumatol. 2009;28:1107-11.
-
15Zancanaro PCQ, Isaac AR, Garcia LT, Costa IMC. Esclerodermia localizada na criança: aspectos clínicos, diagnósticos e terapêuticos. An Bras Dermatol. 2009;84:161-72.
-
16Heilbron B, Saxe N. Scleredema in an infant. Arch Dermatol. 1986;122:1417-19.
-
17Bopp C. Mucinoses cutâneas. An Bras Dermatol. 1968;43:7-24.
Datas de Publicação
-
Publicação nesta coleção
08 Nov 2011 -
Data do Fascículo
Ago 2011
Histórico
-
Recebido
26 Ago 2010 -
Aceito
01 Nov 2010