Abstracts
Several distinct clinical forms of mycosis fungoides have been described. Hypopigmented mycosis fungoides should be regarded as a subtype of mycosis fungoides, insofar as it presents some peculiar characteristics that contrast with the clinical features of the classical form. Most patients with hypopigmented mycosis fungoides are younger than patients typically diagnosed with classical mycosis fungoides. In addition to typical dark-skinned individuals impairment, hypopigmented mycosis fungoides has also been described in Asian patients. The prognosis for hypopigmented mycosis fungoides is much better than for classical mycosis fungoides: hypopigmented mycosis fungoides is diagnosed when there are only patches of affected skin, and lesions usually will not progress beyond terminal stages, although they can persist for many years. Diagnosis should involve clinicopathologic correlation: skin biopsy analysis often reveals intense epidermotropism, characterized by haloed, large, and atypical CD8+ lymphocytes with convoluted nuclei, in contrast to mild to moderate dermal lymphocytic infiltrate. These CD8+ cells, which participate in T helper 1-mediated immune responses, prevent evolution to mycosis fungoides plaques and tumors and could be considered the main cause of the inhibition of melanogenesis. Therefore, hypopigmentation could be considered a marker of good prognosis for mycosis fungoides.
CD8-Positive T-Lymphocytes; Hypopigmentation; Lymphoma, T-Cell, Cutaneous; Melanocytes; Mycosis fungoides
Ultimamente diferentes formas clínicas da micose fungoide têm sido descritas. A micose fungoide hipocromiante pode ser considerada um subtipo da micose fungoide, apresentando algumas características peculiares que contrastam com os achados da forma clássica da micose fungoide. A maioria dos pacientes com micose fungoide hipocromiante são mais jovens que aqueles acometidos pela micose fungoide clássica. Esta variante é descrita principalmente em indivíduos melanodérmicos (afroamericanos e asiáticos). O prognóstico é melhor que o observado para a forma clássica: ao diagnóstico, os pacientes apresentam somente "patches", que tendem a perdurar por longos períodos, sem evolução para estágios mais avançados. O diagnóstico é feito através da correlação clinicopatológica: biópsia da lesão cutânea frequentemente revela intenso epidermotropismo, caracterizado por linfócitos CD8+ atípicos, grandes, com halo e núcleo convoluto, contrastando com o infiltrado dérmico leve a moderado. Estas células CD8+, que participam do perfil de resposta T helper-1, impediriam a evolução da doença para o desenvolvimento de placas infiltradas e tumores, além de determinar a inibição da melanogênese nas lesões hipocrômicas. Portanto, a hipocromia poderia ser considerada um marcador de bom prognóstico na micose fungoide.
Hipopigmentação; Linfoma cutâneo de células T; Linfócitos T CD8-positivos; Melanócitos; Micose fungóide
INTRODUCTION
Mycosis fungoides (MF), the most common primary cutaneous T-cell lymphoma, is a
neoplasic disease characterized by classical non-infiltrated lesions (patches), plaques,
tumors, and erythrodermic stages.11. Yamashita T, Abbade LPF, Marques MEA, Marques SA. Mycosis fungoides
and Sézary syndrome: clinical, histopathological and immunohistochemical review and
update. An Bras Dermatol. 2012;87:817-30.
2. Criscione VD, Weinstock MA.Incidence of cutaneous T-cell lymphoma in
the United States, 1973-2002.Arch Dermatol. 2007;143:854-9.
3. Keehn CA, Belongie IP, Shistik G, Fenske NA, Glass LF.The diagnosis,
staging, and treatment options for mycosis fungoides. Cancer Control.
2007;14:102-11.-44. FestaNeto C, Sanches JA, Melotti CZ. Lymphoproliferative processes of
the skin. Part 2 - Cutaneous T-cell and NK-cell lymphomas. An Bras Dermatol.
2006;87:7-25 Several distinct
clinical forms of MF have been described. These include granulomatous, pustular,
purpuric, hyperkeratotic and verrucous, bullous, invisible, and hypopigmented variants
of the disease.55. Ackerman AB, Miller RC, Shapiro L.Pustular mycosis fungoides.Arch
Dermatol. 1966;93:221-5.
6. Gordon H. Mycosis fungoides. Br J Dermatol
1950;62:177-82.
7. Price NM, Fuks ZY, Hoffman TE.Hyperkeratotic and verrucous features
of mycosis fungoides.Arch Dermatol. 1977;113:57-60.
8. Roenigk HH Jr, Castrovinci AJ.Mycosis fungoidesbullosa.Arch Dermatol.
1971;104:402-6.
9. Pujol RM, Gallardo F, Llistosella E, Blanco A, Bernadó L, Bordes R,
et al. Invisible mycosis fungoides: a diagnostic challenge. J Am AcadDermatol.
2002;47:S168-71.-1010. Ryan EA, Sanderson KV, Barták P, Samman PD. Can mycosis fungoides
begin in the epidermis? A hypothesis.Br J Dermatol. 1973;88:419-29. Zackheim and McCalmont enumerated 25 different diseases that MF
may mimic, referring to it as 'the great imitator'-a description previously reserved for
syphilis.1111. Zackheim HS, McCalmont TH. Mycosis fungoides: the great imitator. J
Am AcadDermatol. 2002;47:914-8.
Hypochromia in MF was previously associated with well-defined clinical forms, such as
poikilodermatous MF or resulting from phototherapy treatment.1212. Choe YB, Park KC, Cho KH.A case of hypopigmented mycosis fungoides.J
Dermatol. 2000;27:543-6. The first case of primary hypopigmented MF (HMF) was described by
Ryan et al. in 1973.1010. Ryan EA, Sanderson KV, Barták P, Samman PD. Can mycosis fungoides
begin in the epidermis? A hypothesis.Br J Dermatol. 1973;88:419-29. Today,
HMF, composed of exclusively hypopigmented or even achromic lesions, can be considered a
subtype of MF, since it presents some peculiar characteristics that contrast with the
clinical features of the classical form. Although HMF has been the subject of several
publications, most of these are case reports or series of cases. There are no criteria
that define a typical case of HMF. Patients with other subtypes of MF and hypochromic
lesions are usually diagnosed as having HMF.1313. Neuhaus IM, Ramos-Caro FA, Hassanein
AM.Hypopigmentedmycosisfungoides in childhoodandadolescense. PediatrDermatol.
2000;17:403-6.
14. Ozcan D, Seçkin D, Ozdemir BH.Hypopigmented macules in na adult male
patient. ClinExpDermatol. 2008;33:667-8.-1515. Ardigó M, Borroni G, Muscardin L, Kerl H, Cerroni L.Hypopigmented
mycosis fungoides in caucasian patients: a clinicopathology study of 7 cases. J
AmAcadDermatol. 2003;49:264-70. The aim of this
review is to justify why hypopigmentation, and no other concomitant type of MF-related
lesion, should be regarded as the defining feature of HMF. We will also discuss the
pathophysiology of HMF and review mechanisms responsible for the hypopigmentation
observed in HMF patients.
Epidemiological findings
Unlike conventional MF, which is regarded as a disease most commonly found in the fifth to sixth decades of life, HMF most commonly affects the pediatric population.33. Keehn CA, Belongie IP, Shistik G, Fenske NA, Glass LF.The diagnosis, staging, and treatment options for mycosis fungoides. Cancer Control. 2007;14:102-11.,1313. Neuhaus IM, Ramos-Caro FA, Hassanein AM.Hypopigmentedmycosisfungoides in childhoodandadolescense. PediatrDermatol. 2000;17:403-6.,1616. Stone ML, Styles AR, Cockerell CJ, Pandya AG.Hypopigmented mycosis fungoides: a report of 7 cases and review of the literature. Cutis. 2001;67:133-8.,1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93. HMF may account for 17% to 59% of all MF cases diagnosed during childhood.1818. Singh ZN, Tretiakova MS, Shea CR, Petronic-Rosic VM. Decreased CD117 expression in hypopigmented mycosis fungoides correlates with hypomelanosis:lessons learned from vitiligo. Mod Pathol. 2006;19:1255-60.,1919. Pope E, Weitzman S, Ngan B, Walsh S, Morel K, Williams J,et al. Mycosis fungoides in the pediatric population: report from an international Childhood Registry of Cutaneous Lymphoma. J Cutan Med Surg. 2010;14:1-6. An analysis of 131 new cases of MF/Sézary syndrome (SS) in Singapore over a 5-year period reported a median age of 33 at the time of diagnosis, with a mean age of 36.3.2020. Tan ES, Tang MB, Tan SH.Retrospective 5-year review of 131 patients with mycosis fungoides and Sézary syndrome seen at the National Skin Centre, Singapore.Australas J Dermatol. 2006;47:248-52. However, the median age of the 47 patients within the cohort who were diagnosed with the HMF variant of MF/SS was only 17 at the time of diagnosis.2020. Tan ES, Tang MB, Tan SH.Retrospective 5-year review of 131 patients with mycosis fungoides and Sézary syndrome seen at the National Skin Centre, Singapore.Australas J Dermatol. 2006;47:248-52. Another study, which encompassed almost the entire population of Kuwait and included 193 cases of MF, identified 77 patients with hypopigmented macules, alone or in combination with other types of skin lesions.2121. Alsaleh QA, Nanda A, Al-Ajmi H, Al-Sabah H, Elkashlan M, Al-Shemmari S, et al. Clinicoepidemiological features of mycosis fungoides in Kuwait, 1991-2006. Int J Dermatol. 2010;49:1393-8. For 43 patients (22% of the entire sample) with the pure hypopigmented variant, the mean age at the time of diagnosis (27.6 ± 12.42) was younger than for all other MF cases (38.14 ± 14.37). Previous data from the literature also confirm the relatively young age of onset of HMF, in contrast to the typical age of population affected by classical MF.1515. Ardigó M, Borroni G, Muscardin L, Kerl H, Cerroni L.Hypopigmented mycosis fungoides in caucasian patients: a clinicopathology study of 7 cases. J AmAcadDermatol. 2003;49:264-70.,1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93.
A predilection for individuals with high phototypes (Fitzpatrick scale of skin types)
is also a peculiar manifestation of HMF. Accordingly, HMF is reported almost
exclusively in dark-skinned and Asian patients. Classical MF is also found more
frequently in individuals with dark skin.33. Keehn CA, Belongie IP, Shistik G, Fenske NA, Glass LF.The diagnosis,
staging, and treatment options for mycosis fungoides. Cancer Control.
2007;14:102-11. The
classical form of MF is by far the most common form of MF in dark-skinned
individuals, with HMF regarded merely as an atypical and rare variant for these
populations.1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo
D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and
treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93.,2222. Moulonguet I, Robert C, Baudot N, Flageul B, Dubertret
L.Hypopigmented mycosis fungoides in a light-skinned woman.Br J Dermatol.
1998;139:341-3.
23. Akaraphanth R, Douglass MC, Lim HW.Hypopigmented mycosis fungoides:
treatment and a 6 1/2 -year follow-up of 9 patients. J Am AcadDermatol.
2000;42:33-9.-2424. Halder RM, Ara CJ. Skin câncer and photoaging in ethnic
skin.DermatolClin. 2003;21:725-32, x. Asians are commonly described as presenting HMF, although classical MF
is not very prevalent among Asian populations.33. Keehn CA, Belongie IP, Shistik G, Fenske NA, Glass LF.The diagnosis,
staging, and treatment options for mycosis fungoides. Cancer Control.
2007;14:102-11.,2525. Das JK, Gangopadhyay AK.Mycosis fungoides with unsualvitiligo-like
presentation.Indian J DermatolVenereolLeprol. 2004;70:304-6.
26. Hsiao PF, Hsiao CH, Tsai TF, Jee SH.Minimal reisdual disease in
hypopigmented mycosis fungoides.J Am AcadDermatol. 2006;54:S198-201.-2727. Tan E, Tay YK, Giam YC.Profile and outcome of childhood mycosis
fungoides in Singapore.Pediatr Dermatol. 2000;17:352-6. Few cases of HMF have been reported in
Caucasians.2828. Hodak E, Phenig E, Amichai B, Feinmesser M, Kuten A, Maron L,et al.
Unilesionalmycosisfungoides. Dermatology. 2000;201:300-6. Of seven Caucasian patients
diagnosed with HMF, five had hypopigmented patches, in addition to erythematous
lesions.1515. Ardigó M, Borroni G, Muscardin L, Kerl H, Cerroni L.Hypopigmented
mycosis fungoides in caucasian patients: a clinicopathology study of 7 cases. J
AmAcadDermatol. 2003;49:264-70. It was concluded that this
"mixed" clinical pattern could be predominant in Caucasian patients.
Although it is generally considered that there is no gender predilection for HMF, some authors, such as Shabrawi-Caelen et al., observed a striking female predominance in a study involving 11 females and only 4 males.1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93.,2020. Tan ES, Tang MB, Tan SH.Retrospective 5-year review of 131 patients with mycosis fungoides and Sézary syndrome seen at the National Skin Centre, Singapore.Australas J Dermatol. 2006;47:248-52.,2929. El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH.Hypopigmented mycosis fungoides.Frequent expression of a CD8+ T-cell phenotype.Am J SurgPathol. 2002;26:450-7. This contrasts with the reality of classical MF, which is more prevalent in males.11. Yamashita T, Abbade LPF, Marques MEA, Marques SA. Mycosis fungoides and Sézary syndrome: clinical, histopathological and immunohistochemical review and update. An Bras Dermatol. 2012;87:817-30.
The frequency of HMF is unknown, although most authors agree that it is underestimated. Misdiagnosis of HMF as any of a range of differential diagnoses, including vitiligo, pityriasis alba, leprosy, and postinflammatory hypopigmentation, is common.1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93. The limited number of publications on HMF has signified poor knowledge of HMF for many professionals, thus preventing correct diagnosis.
Clinical findings
The defining clinical features of HMF are hypopigmented-to-achromic lesions, sometimes with a vitiligo-like aspect, which are mainly distributed on the trunk and proximal portions of the extremities, especially the buttocks, as well as the pelvic girdle and the lower limbs.11. Yamashita T, Abbade LPF, Marques MEA, Marques SA. Mycosis fungoides and Sézary syndrome: clinical, histopathological and immunohistochemical review and update. An Bras Dermatol. 2012;87:817-30.,2525. Das JK, Gangopadhyay AK.Mycosis fungoides with unsualvitiligo-like presentation.Indian J DermatolVenereolLeprol. 2004;70:304-6.,3030. Werner B, Brown S, Ackerman AB.Hypopigmented mycosis fungoides is not always mycosis fungoides! Am J Dermatopathol. 2005;27:56-67. Variations of these tendencies include involvement of the distal extremities, such as the head and neck.2626. Hsiao PF, Hsiao CH, Tsai TF, Jee SH.Minimal reisdual disease in hypopigmented mycosis fungoides.J Am AcadDermatol. 2006;54:S198-201.,2727. Tan E, Tay YK, Giam YC.Profile and outcome of childhood mycosis fungoides in Singapore.Pediatr Dermatol. 2000;17:352-6. Patch sizes vary, ranging from droplet-size to large plate-size lesions.1414. Ozcan D, Seçkin D, Ozdemir BH.Hypopigmented macules in na adult male patient. ClinExpDermatol. 2008;33:667-8. Patients have ever been related with a single lesion.2828. Hodak E, Phenig E, Amichai B, Feinmesser M, Kuten A, Maron L,et al. Unilesionalmycosisfungoides. Dermatology. 2000;201:300-6.,3131. Roupe G. Hypopigmented mycosis fungoides in a child successfully treated with UVA-1 light. Pediatr Dermatol. 2005;22:82. Patients may complain of itchiness, with variable intensity, although local sensitivity is always preserved.1313. Neuhaus IM, Ramos-Caro FA, Hassanein AM.Hypopigmentedmycosisfungoides in childhoodandadolescense. PediatrDermatol. 2000;17:403-6.,3232. Manzur A, Zaidi ST.Hypopigmented mycosis fungoides in a 10 year-old boy.Dermatol Online J. 2006;12:21.,3333. Goldberg DJ, Schinella RS, Kechijian P.Hypopigmented mycosis fungoides.Speculations about the mechanism of hypopigmentation. Am J Dermatopathol. 1986;8:326-30. Other findings include lesions with atrophy and telangiectasia, and lesions that become more evident after sun exposure.2222. Moulonguet I, Robert C, Baudot N, Flageul B, Dubertret L.Hypopigmented mycosis fungoides in a light-skinned woman.Br J Dermatol. 1998;139:341-3.,3434. Hsu CK, Hsu MM, Lee JY.Fusariosisoccuring in a ulcerated cutaneous CD8+ T cell lymphoma tumor. Eur J Dermatol. 2006;16:297-301. These are described in figures 1-3.
Usual clinical presentation: typical hypopigmented patches on chest (A), abdomen (B) and right buttock and thigh (C)
Unusual forms of hypopigmented patches: guttata (A) – buttocks and thighs, and vitiligoid lesions (B) – left thigh
Differential diagnosis should include atopic dermatitis, pityriasis alba, leprosy, vitiligo, postinflammatory hypopigmentation, sarcoidosis, pityriasis lichenoides chronica, pityriasis versicolor, syphilis and other treponematosis, idiophatic guttate hypomelanosis, lichen sclerosus, hypomelanosis of Ito, halo nevus, and onchocerciasis.1313. Neuhaus IM, Ramos-Caro FA, Hassanein AM.Hypopigmentedmycosisfungoides in childhoodandadolescense. PediatrDermatol. 2000;17:403-6.,1414. Ozcan D, Seçkin D, Ozdemir BH.Hypopigmented macules in na adult male patient. ClinExpDermatol. 2008;33:667-8.,1616. Stone ML, Styles AR, Cockerell CJ, Pandya AG.Hypopigmented mycosis fungoides: a report of 7 cases and review of the literature. Cutis. 2001;67:133-8.,2929. El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH.Hypopigmented mycosis fungoides.Frequent expression of a CD8+ T-cell phenotype.Am J SurgPathol. 2002;26:450-7. Undoubtedly, in regions where leprosy is endemic, HMF can be easily misdiagnosed as mycobacteriosis. Like some authors, we have encountered several cases of HMF in which patients had previously received treatment for leprosy (unpublished data).3535. Khopkar U, Doshi BR, Dongre AM, Gujral S.A study of clinicopathologic profile of 15 cases of hypopigmented mycosis fungoides.Indian J Dermatol Venereol Leprol. 2011;77:167-73.
The prognosis for MFH is usually excellent, at least when compared with classical MF/SS. Most HMF patients are diagnosed during early stages of the disease, when there are only patches, without infiltrated plaques.2222. Moulonguet I, Robert C, Baudot N, Flageul B, Dubertret L.Hypopigmented mycosis fungoides in a light-skinned woman.Br J Dermatol. 1998;139:341-3.,2323. Akaraphanth R, Douglass MC, Lim HW.Hypopigmented mycosis fungoides: treatment and a 6 1/2 -year follow-up of 9 patients. J Am AcadDermatol. 2000;42:33-9. This presentation tends to last for many years and does not progress beyond stage IB (defined by the International Society of Cutaneous Lymphomas as lesions covering >10% body surface area without involvement of lymph nodes or the viscera). Notwithstanding the better prognosis for HMF patients compared with classical MF patients, the same authors reported patient deaths resulting from this cutaneous lymphoma.1515. Ardigó M, Borroni G, Muscardin L, Kerl H, Cerroni L.Hypopigmented mycosis fungoides in caucasian patients: a clinicopathology study of 7 cases. J AmAcadDermatol. 2003;49:264-70.,1616. Stone ML, Styles AR, Cockerell CJ, Pandya AG.Hypopigmented mycosis fungoides: a report of 7 cases and review of the literature. Cutis. 2001;67:133-8. Thus, despite the good prognosis, HMF should always be treated as a malignant neoplastic disease, and its potential lethality should never be underestimated. Clinical assessment of patients with this variant is always necessary, with complete staging that should include peripheral blood examination, quantification of Sézary cells and T lymphocytes using flow cytometry, physical examination of the peripheral lymph nodes (with biopsy if their diameter widens to > 1.5 cm), and imaging studies to exclude visceral impairment.3636. Olsen EA, Whittaker S, Kim YH, Duvic M, Prince HM, Lessin SR, et al. Clinical end points and response criteria in mycosis fungoides and Sézary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011;29:2598-607.
DIAGNOSIS
Clinicopathologic correlation should be used to diagnose HMF. Even if the histological evidence from one or more skin samples is inadequate to make a conclusive diagnosis of HMF, the patient should be closely followed-up as potentially having HMF. Immunohistochemistry provides a very valuable complementary tool, since a typical CD8 immunophenotype could be considered a hallmark of HMF. Ultrastructural analysis, using electron microscopy and molecular techniques, can also be useful, although these may not be available in clinical practice.
The typical histopathological features of HMF are identical to those of all other patch and plaque variants of MF. A review of 106 cases reported in 33 publications defined some of the most common changes as focal parakeratosis, little or no spongiosis, lymphocytic infiltrate in the upper dermis where some bundles of collagen seemed to be coarse, and a variable number of lymphocytes at all levels of the viable epidermis that are disposed as solitary units and, episodically, in tiny collections.3030. Werner B, Brown S, Ackerman AB.Hypopigmented mycosis fungoides is not always mycosis fungoides! Am J Dermatopathol. 2005;27:56-67. Several authors have reported very intense epidermotropism, characterized by haloed, large, and atypical lymphocytes with convoluted nuclei, in contrast to mild to moderate dermal lymphocytic infiltrate.1212. Choe YB, Park KC, Cho KH.A case of hypopigmented mycosis fungoides.J Dermatol. 2000;27:543-6.,1313. Neuhaus IM, Ramos-Caro FA, Hassanein AM.Hypopigmentedmycosisfungoides in childhoodandadolescense. PediatrDermatol. 2000;17:403-6.,2222. Moulonguet I, Robert C, Baudot N, Flageul B, Dubertret L.Hypopigmented mycosis fungoides in a light-skinned woman.Br J Dermatol. 1998;139:341-3. These findings (intense epidermotropism) are not routinely observed in early lesions of classical MF. Pautrier microabscesses are seldom described. Other changes associated with HMF include slight psoriasiform epidermal hyperplasia, scattered dyskeratotic keratinocytes, vacuolar alteration of the dermoepidermal junction simulating an interface dermatitis, folliculotropism, and melanin incontinence with melanophages in the papillary dermis.2929. El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH.Hypopigmented mycosis fungoides.Frequent expression of a CD8+ T-cell phenotype.Am J SurgPathol. 2002;26:450-7. Dermal infiltrate from HMF patients is described as sparse superficial perivascular or patchy lichenoid.1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93.,3535. Khopkar U, Doshi BR, Dongre AM, Gujral S.A study of clinicopathologic profile of 15 cases of hypopigmented mycosis fungoides.Indian J Dermatol Venereol Leprol. 2011;77:167-73. However, histological findings may not be conclusive and repeated biopsies are required during follow-ups to confirm the diagnosis. Histopatholo-gical findings are illustrated in figure 4.
Histopathologic findings. This skin biopsy section shows parakeratosis (a), lymphocytes at all levels of the epidermis, including the basal cell layer (b) and upper dermis lymphocytic infiltrate (c)
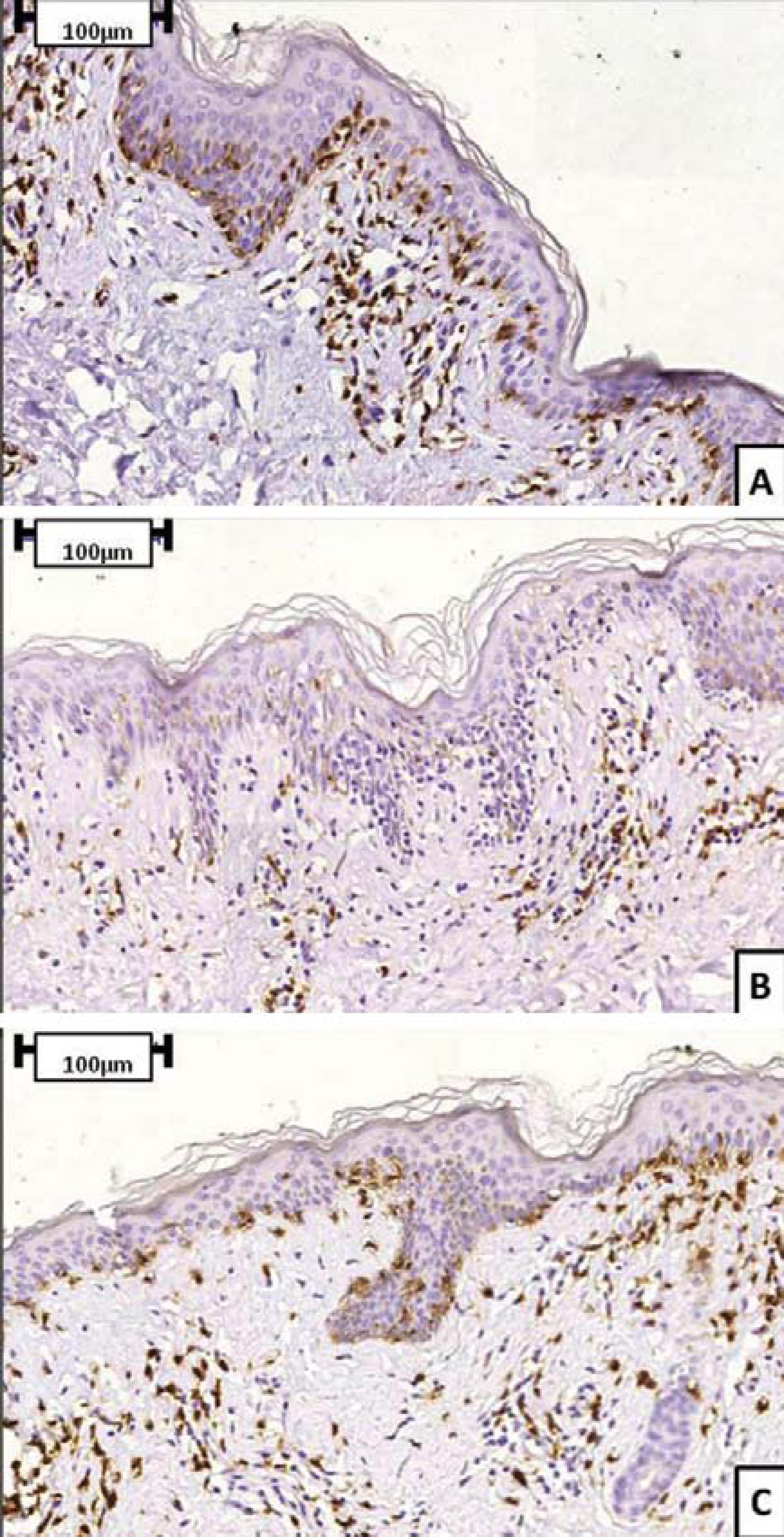
Immunohistochemical analysis can reveal peculiarities that allow HMF to be differentiated from classical MF. Epidermotropism in classical MF is characterized by neoplastic CD4+ T cells, with CD8+ T cells representing a minority of the reactive T lymphocytes and residing predominantly in the dermal papillae. Neoplastic CD8+ T-cell infiltrate is very uncommon.3535. Khopkar U, Doshi BR, Dongre AM, Gujral S.A study of clinicopathologic profile of 15 cases of hypopigmented mycosis fungoides.Indian J Dermatol Venereol Leprol. 2011;77:167-73. However, in HMF, several authors have shown a predominant CD8+ cell epidermotropism.1313. Neuhaus IM, Ramos-Caro FA, Hassanein AM.Hypopigmentedmycosisfungoides in childhoodandadolescense. PediatrDermatol. 2000;17:403-6.,1515. Ardigó M, Borroni G, Muscardin L, Kerl H, Cerroni L.Hypopigmented mycosis fungoides in caucasian patients: a clinicopathology study of 7 cases. J AmAcadDermatol. 2003;49:264-70.,1818. Singh ZN, Tretiakova MS, Shea CR, Petronic-Rosic VM. Decreased CD117 expression in hypopigmented mycosis fungoides correlates with hypomelanosis:lessons learned from vitiligo. Mod Pathol. 2006;19:1255-60.,2929. El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH.Hypopigmented mycosis fungoides.Frequent expression of a CD8+ T-cell phenotype.Am J SurgPathol. 2002;26:450-7.,3737. Wain EM, Orchard GE, Whittaker SJ, Spittle M Sc MF, Russell-Jones R.Outcome in 34 patients with juvenile-onset mycosis fungoides.Cancer. 2003;98:2282-90.,3838. El-Darouti MA, Marzouk SA, Azzam O, Fawzi MM, Abdel-Halim MR, Zayed AA, et al. Vitiligo vs. Hypopigmented mycosis fungoides (histopathological and immunohistochemical study, univariate analysis). Eur J Dermatol. 2006;16:17-22. A CD8+ immunophenotype case is presented in figure 5. On the other hand, some papers have confirmed that HMF lesions can present typical features of classical MF, such as a predominance of CD4+ T cells and preservation of a normal ratio of CD4+ lymphocytes to CD8+ lymphocytes.1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93. The prevalence of CD8+ T cells can influence significantly the pathogenesis of HMF, given that the suppressor phenotype seems to limit progression of the disease by impairing cutaneous dissemination and preventing the onset of a more aggressive stage, despite the neoplastic nature of these cells.1616. Stone ML, Styles AR, Cockerell CJ, Pandya AG.Hypopigmented mycosis fungoides: a report of 7 cases and review of the literature. Cutis. 2001;67:133-8.,3939. Volkenandt M, Soyer HP, Cerroni L, Koch OM, Atzpodien J, Kerl H.Molecular detection of clone-specific DNA in hypopigmented lesions of a patient with early evolving mycosis fungoides.Br J Dermatol. 1993;128:423-8. A similar role may be played by reactive CD8+ T cells if the infiltrate is characterized by a prevalent number of CD4+ cells. A few studies have analyzed rearrangements of the genes encoding T-cell receptors (TCR) during progression of HMF. For instance, El- ShabrawiCaelenet al.2929. El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH.Hypopigmented mycosis fungoides.Frequent expression of a CD8+ T-cell phenotype.Am J SurgPathol. 2002;26:450-7. demonstrated TCR-gene rearrangement in 6 of 13 patients with HMF. Evidence of monoclonal gene rearrangement in intraepidermal lymphocytes was noted in five of these six patients. The authors demonstrated that the malignant population in 5 out of 7 patients was derived from neoplastic CD8+ T cells that resided either within the epidermis or the lining at the dermoepidermal junction.2929. El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH.Hypopigmented mycosis fungoides.Frequent expression of a CD8+ T-cell phenotype.Am J SurgPathol. 2002;26:450-7. Other authors found no TCR gene rearrangement and attributed this absence to the early stage and/or the "benign" nature of HMF.1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93.,2222. Moulonguet I, Robert C, Baudot N, Flageul B, Dubertret L.Hypopigmented mycosis fungoides in a light-skinned woman.Br J Dermatol. 1998;139:341-3.,4040. Di Landro A, Marchesi L, Naldi L, Motta T, Cainelli T.A case of hypopigmented mycosis fungoides in a Young caucasian boy. Pediatr Dermatol. 1997;14:449-52. Larger series are required to make more conclusive statements regarding the role of TCR gene rearrangement in the progression of HMF.
Immunohistochemical staining (CD3, CD4 and CD8) of a HMF case. Sequence represents CD3 (A), CD4 (B) and CD8 (C) staining. This patient presented a predominance of CD8+ over CD4+ cells in neoplastic epidermotropism)
PATHOPHYSIOLOGY
Ultrastructural analysis, using electron microscopy, suggested that atypical neoplastic cells could cause degeneration of melanocytes and abnormal melanogenesis.4141. Breathnach SM, McKee PH, Smith NP.Hypopigmented mycosis fungoides: report of five cases with ultrastructural observations. Br J Dermatol. 1982;106:643-9. Given the absence of evidence that melanosome transfer was blocked, the authors concluded that changes were a nonspecific response of cell injury associated with inflammation.4141. Breathnach SM, McKee PH, Smith NP.Hypopigmented mycosis fungoides: report of five cases with ultrastructural observations. Br J Dermatol. 1982;106:643-9. Another study involving electron microscopy demonstrated a reduced number of normal-appearing melanosomes within keratinocytes of patients with HMF, despite finding abundant and morphologically normal melanosomes in adjacent normal melanocytes.3333. Goldberg DJ, Schinella RS, Kechijian P.Hypopigmented mycosis fungoides.Speculations about the mechanism of hypopigmentation. Am J Dermatopathol. 1986;8:326-30. These melanocytes did not present degenerative changes. After treatment with topical nitrogen mustard, electron microscopy of repigmented normal-appearing skin revealed numerous normal-appearing melanosomes within keratinocytes. It was thus concluded that hypopigmentation of HMF may arise from a defect in the transfer of melanosomes from melanocytes to keratinocytes, with reversal of this defect after treatment.3333. Goldberg DJ, Schinella RS, Kechijian P.Hypopigmented mycosis fungoides.Speculations about the mechanism of hypopigmentation. Am J Dermatopathol. 1986;8:326-30. Several other studies have shown either fewer melanocytes and altered melanocyte morphology, or defective transfer of melanosomes.1313. Neuhaus IM, Ramos-Caro FA, Hassanein AM.Hypopigmentedmycosisfungoides in childhoodandadolescense. PediatrDermatol. 2000;17:403-6.,1616. Stone ML, Styles AR, Cockerell CJ, Pandya AG.Hypopigmented mycosis fungoides: a report of 7 cases and review of the literature. Cutis. 2001;67:133-8.,2222. Moulonguet I, Robert C, Baudot N, Flageul B, Dubertret L.Hypopigmented mycosis fungoides in a light-skinned woman.Br J Dermatol. 1998;139:341-3.,4242. Bouloc A, Grange F, Delfau-Larue MH, Dieng MT, Tortel MC, Avril MF, et al. Leucoderma associated with flares of erythrodermic cutaneous T-cell lymphomas:four cases. Br J Dermatol. 2000;143:832-6. However, it is possible that these two assumptions are valid, with abnormal melanogenesis comprising a continuum, on which melanocyte death is the most extreme expression of pathology.1717. Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93.
A predominance of CD8+ T cells in neoplastic infiltrates is consistent with the hypothesis that hypopigmentation results from the cytotoxicity of suppressor lymphocytes towards melanocytes. It is unclear whether non-neoplastic reactive CD8+ T cells could play a similar role in lesions for which the infiltrate is characterized by a predominance of CD4+ T cells.1515. Ardigó M, Borroni G, Muscardin L, Kerl H, Cerroni L.Hypopigmented mycosis fungoides in caucasian patients: a clinicopathology study of 7 cases. J AmAcadDermatol. 2003;49:264-70.,2525. Das JK, Gangopadhyay AK.Mycosis fungoides with unsualvitiligo-like presentation.Indian J DermatolVenereolLeprol. 2004;70:304-6.,4343. Chuang GS, Wasserman DI, Byers HR, Demierre MF.Hypopigmented T-cell discrasia evolving to hypopigmented mycosis fungoides during etanercept therapy.J Am AcadDermatol. 2008;59:S121-2. Hence, neoplastic and non-neoplastic lymphocytes might be able to inhibit or even destroy melanocytes from affected skin, causing hypopigmented lesions.
Initially, MF patches present a predominant T helper 1 (Th1)-type immune response, with substantial participation of interferon-gamma (IFNγ) and CD8+ cells. Evolution of the pathology to infiltrated plaques and tumors is characterized by a shift towards predominance of a T helper 2 (Th2)-type immune response. Thus, the Th1 response is seen as a pathway of defense against cancer, and a change in the immune pattern (from Th1 to Th2) is associated with a worse prognosis.4444. Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM,et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798-812. The same neoplastic behaviors likely apply as much to HMF as they do to classical MF.
Hypopigmentation could represent the extreme result of a protective immune response: neoplastic or reactive CD8+ T cells in cellular infiltrates guarantee an indolent disease with good prognosis, and prevent transformation to a Th2-type immune response, which is associated with progression to malignancy.
Studies comparing mechanisms of hypopigmentation of vitiligo and HMF have made an important contribution to the understanding of their pathogeneses. Moretti et al.4545. Moretti S, Spallanzani A, Amato L, Hautmann G, Gallerani I, Fabiani M, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92. demonstrated significant changes in the levels of different epidermal cytokines in vitiligo lesions, in which there were no melanocytes. Within the lesions, levels of cytokines with stimulatory activities towards melanocytes (e.g., granulocyte-macrophage colony-stimulating factor, stem cell factor, and basic fibroblast growth factor bFGF) were reduced, whereas levels of cytokines with inhibitory activities (e.g., interleukin-6 and tumor necrosis factor α- TNF-α) were significantly increased. These results seem to confirm, at least in part, the inhibition of melanogenesis induced by TNFα in vitro, through inhibition of melanocytic tyrosinase and tyrosinase-related protein 1, both of which are essential for melanin synthesis.4545. Moretti S, Spallanzani A, Amato L, Hautmann G, Gallerani I, Fabiani M, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92. Recently, Seif El Nasr et al.4646. Seif El Nasr H, Shaker OG, Fawzi MM, El-Hanafi G. Basic fibroblast growth factor and tumour necrosis factor alpha in vitiligo and other hypopigmented disorders: suggestive possible therapeutic targets. J EurAcadDermatolVenereol. 2013;27:103-8. showed a similar increase in TNF-α levels and decreased expression of mRNA encoding bFGF in HMF lesions. Vitiligo lesions are characterized by a reduction in expression of the CD117 receptor (also known as c-KIT, a stem cell factor receptor that regulates melanocyte activity and stability) in epidermal melanocytes, which increases apoptosis rates in these cells.1818. Singh ZN, Tretiakova MS, Shea CR, Petronic-Rosic VM. Decreased CD117 expression in hypopigmented mycosis fungoides correlates with hypomelanosis:lessons learned from vitiligo. Mod Pathol. 2006;19:1255-60. Vitiligo lesions also have a predominance of epidermal CD8+ T-cell infiltrate. Similar changes were found in HMF, but not in classical MF lesions or normal skin. Thus, cytotoxic effects of CD8+ T cells on HMF lesions presumably lead to a decreased expression of CD117 in melanocytes, resulting in dysfunction and/or loss of these cells.1818. Singh ZN, Tretiakova MS, Shea CR, Petronic-Rosic VM. Decreased CD117 expression in hypopigmented mycosis fungoides correlates with hypomelanosis:lessons learned from vitiligo. Mod Pathol. 2006;19:1255-60. Kitamura et al. demonstrated that keratinocytes on the edges of vitiligo lesions secrete elevated levels of two melanocyte stimulating cytokines, stem cell factor (the ligand of CD117) and endothelin.4747. Kitamura R, Tsukamoto K, Harada K, Shimizu A, Shimada S, Kobayashi T, et al. Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis: role of SCF/KIT protein interactions and the downstream effector, MITF-M. J Pathol. 2004;202:463-75. These changes were accompanied by a decrease in secretion rates in CD117 and the microphthalmia-associated transcription factor (MITF), which interacts with CD117 to control directly melanin synthesis and melanocyte survival. No change in the expression of the ET-receptor 1 was noted.4747. Kitamura R, Tsukamoto K, Harada K, Shimizu A, Shimada S, Kobayashi T, et al. Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis: role of SCF/KIT protein interactions and the downstream effector, MITF-M. J Pathol. 2004;202:463-75. Thus, decreased levels of CD117 and MITF are associated with increased rates of melanocyte apoptosis. In addition, CD117 keeps melanocytes within the epidermis, through affinity for stem cell factor secreted by keratinocytes.4747. Kitamura R, Tsukamoto K, Harada K, Shimizu A, Shimada S, Kobayashi T, et al. Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis: role of SCF/KIT protein interactions and the downstream effector, MITF-M. J Pathol. 2004;202:463-75.
In summary, the Th1 response, especially that contributed either by neoplastic or reactive CD8+ cells, prevents the evolution of hypopigmented HMF lesions to MF plaques and tumors, and inhibits melanogenesis. Therefore, hypopigmentation should be considered a potential marker of good prognosis.
TREATMENT
Phototherapy, especially photochemotherapy, is the most widely used method.4848. Duarte I, Bedrikow R, Aoki S. Mycosis fungoides: epidemiologic study of 17 cases and evaluation of PUVA photochemotherapy. An Bras Dermatol 2006;81:40-5 It ensures rapid and complete remission in most cases, and is recommended as first-line treatment for HMF.33. Keehn CA, Belongie IP, Shistik G, Fenske NA, Glass LF.The diagnosis, staging, and treatment options for mycosis fungoides. Cancer Control. 2007;14:102-11.,1212. Choe YB, Park KC, Cho KH.A case of hypopigmented mycosis fungoides.J Dermatol. 2000;27:543-6.,2727. Tan E, Tay YK, Giam YC.Profile and outcome of childhood mycosis fungoides in Singapore.Pediatr Dermatol. 2000;17:352-6. Narrow-band ultraviolet radiation has also been widely used, especially in children.4949. Onsun N, Kural Y, Su O, Demirkesen C, Büyükbabani N.Hypopigmented mycosis fungoides associated with atopy in two children. PediatrDermatol. 2006;23:493-6. Although a poor response to narrow-band ultraviolet radiation has been described for dark-skinned individuals, possibly owing to a photoprotective effect of melanin, some authors have reported its effectiveness in Asian patients.3232. Manzur A, Zaidi ST.Hypopigmented mycosis fungoides in a 10 year-old boy.Dermatol Online J. 2006;12:21.,5050. Kanokrungsee S, Rajatanavin N, Rutnin S, Vachiramon V.Efficacy of narrowband ultraviolet B twice weekly for hypopigmented mycosis fungoides in Asians.ClinExpDermatol. 2012;37:149-52. Other effective therapeutic options described are topical nitrogen mustard, topical carmustine, and total skin electron beam therapy.1212. Choe YB, Park KC, Cho KH.A case of hypopigmented mycosis fungoides.J Dermatol. 2000;27:543-6. Given that HMF is a highly recurrent disease, aggressive treatment is contraindicated. Recurrences can occur a few months or even many years after total remission and therefore followup is mandatory.2626. Hsiao PF, Hsiao CH, Tsai TF, Jee SH.Minimal reisdual disease in hypopigmented mycosis fungoides.J Am AcadDermatol. 2006;54:S198-201.,3535. Khopkar U, Doshi BR, Dongre AM, Gujral S.A study of clinicopathologic profile of 15 cases of hypopigmented mycosis fungoides.Indian J Dermatol Venereol Leprol. 2011;77:167-73.
CONCLUSION
HMF is a cutaneous lymphoma found more frequently in young and dark-skinned individuals. Although the prognosis for HMF is generally good, HMF is a malignant skin tumor and should always be treated as such. Follow-up is mandatory and aggressive treatment should be avoided in light of the high rate of recurrence after therapy.
Acknowledgments
We would like to acknowledge the contributions made by Prof. Valéria Aoki, who reviewed a draft of this article.
REFERENCES
-
1Yamashita T, Abbade LPF, Marques MEA, Marques SA. Mycosis fungoides and Sézary syndrome: clinical, histopathological and immunohistochemical review and update. An Bras Dermatol. 2012;87:817-30.
-
2Criscione VD, Weinstock MA.Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002.Arch Dermatol. 2007;143:854-9.
-
3Keehn CA, Belongie IP, Shistik G, Fenske NA, Glass LF.The diagnosis, staging, and treatment options for mycosis fungoides. Cancer Control. 2007;14:102-11.
-
4FestaNeto C, Sanches JA, Melotti CZ. Lymphoproliferative processes of the skin. Part 2 - Cutaneous T-cell and NK-cell lymphomas. An Bras Dermatol. 2006;87:7-25
-
5Ackerman AB, Miller RC, Shapiro L.Pustular mycosis fungoides.Arch Dermatol. 1966;93:221-5.
-
6Gordon H. Mycosis fungoides. Br J Dermatol 1950;62:177-82.
-
7Price NM, Fuks ZY, Hoffman TE.Hyperkeratotic and verrucous features of mycosis fungoides.Arch Dermatol. 1977;113:57-60.
-
8Roenigk HH Jr, Castrovinci AJ.Mycosis fungoidesbullosa.Arch Dermatol. 1971;104:402-6.
-
9Pujol RM, Gallardo F, Llistosella E, Blanco A, Bernadó L, Bordes R, et al. Invisible mycosis fungoides: a diagnostic challenge. J Am AcadDermatol. 2002;47:S168-71.
-
10Ryan EA, Sanderson KV, Barták P, Samman PD. Can mycosis fungoides begin in the epidermis? A hypothesis.Br J Dermatol. 1973;88:419-29.
-
11Zackheim HS, McCalmont TH. Mycosis fungoides: the great imitator. J Am AcadDermatol. 2002;47:914-8.
-
12Choe YB, Park KC, Cho KH.A case of hypopigmented mycosis fungoides.J Dermatol. 2000;27:543-6.
-
13Neuhaus IM, Ramos-Caro FA, Hassanein AM.Hypopigmentedmycosisfungoides in childhoodandadolescense. PediatrDermatol. 2000;17:403-6.
-
14Ozcan D, Seçkin D, Ozdemir BH.Hypopigmented macules in na adult male patient. ClinExpDermatol. 2008;33:667-8.
-
15Ardigó M, Borroni G, Muscardin L, Kerl H, Cerroni L.Hypopigmented mycosis fungoides in caucasian patients: a clinicopathology study of 7 cases. J AmAcadDermatol. 2003;49:264-70.
-
16Stone ML, Styles AR, Cockerell CJ, Pandya AG.Hypopigmented mycosis fungoides: a report of 7 cases and review of the literature. Cutis. 2001;67:133-8.
-
17Lambroza E, Cohen SR, Phelps R, Lebwohl M, Braverman IM, DiCostanzo D.Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-93.
-
18Singh ZN, Tretiakova MS, Shea CR, Petronic-Rosic VM. Decreased CD117 expression in hypopigmented mycosis fungoides correlates with hypomelanosis:lessons learned from vitiligo. Mod Pathol. 2006;19:1255-60.
-
19Pope E, Weitzman S, Ngan B, Walsh S, Morel K, Williams J,et al. Mycosis fungoides in the pediatric population: report from an international Childhood Registry of Cutaneous Lymphoma. J Cutan Med Surg. 2010;14:1-6.
-
20Tan ES, Tang MB, Tan SH.Retrospective 5-year review of 131 patients with mycosis fungoides and Sézary syndrome seen at the National Skin Centre, Singapore.Australas J Dermatol. 2006;47:248-52.
-
21Alsaleh QA, Nanda A, Al-Ajmi H, Al-Sabah H, Elkashlan M, Al-Shemmari S, et al. Clinicoepidemiological features of mycosis fungoides in Kuwait, 1991-2006. Int J Dermatol. 2010;49:1393-8.
-
22Moulonguet I, Robert C, Baudot N, Flageul B, Dubertret L.Hypopigmented mycosis fungoides in a light-skinned woman.Br J Dermatol. 1998;139:341-3.
-
23Akaraphanth R, Douglass MC, Lim HW.Hypopigmented mycosis fungoides: treatment and a 6 1/2 -year follow-up of 9 patients. J Am AcadDermatol. 2000;42:33-9.
-
24Halder RM, Ara CJ. Skin câncer and photoaging in ethnic skin.DermatolClin. 2003;21:725-32, x.
-
25Das JK, Gangopadhyay AK.Mycosis fungoides with unsualvitiligo-like presentation.Indian J DermatolVenereolLeprol. 2004;70:304-6.
-
26Hsiao PF, Hsiao CH, Tsai TF, Jee SH.Minimal reisdual disease in hypopigmented mycosis fungoides.J Am AcadDermatol. 2006;54:S198-201.
-
27Tan E, Tay YK, Giam YC.Profile and outcome of childhood mycosis fungoides in Singapore.Pediatr Dermatol. 2000;17:352-6.
-
28Hodak E, Phenig E, Amichai B, Feinmesser M, Kuten A, Maron L,et al. Unilesionalmycosisfungoides. Dermatology. 2000;201:300-6.
-
29El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH.Hypopigmented mycosis fungoides.Frequent expression of a CD8+ T-cell phenotype.Am J SurgPathol. 2002;26:450-7.
-
30Werner B, Brown S, Ackerman AB.Hypopigmented mycosis fungoides is not always mycosis fungoides! Am J Dermatopathol. 2005;27:56-67.
-
31Roupe G. Hypopigmented mycosis fungoides in a child successfully treated with UVA-1 light. Pediatr Dermatol. 2005;22:82.
-
32Manzur A, Zaidi ST.Hypopigmented mycosis fungoides in a 10 year-old boy.Dermatol Online J. 2006;12:21.
-
33Goldberg DJ, Schinella RS, Kechijian P.Hypopigmented mycosis fungoides.Speculations about the mechanism of hypopigmentation. Am J Dermatopathol. 1986;8:326-30.
-
34Hsu CK, Hsu MM, Lee JY.Fusariosisoccuring in a ulcerated cutaneous CD8+ T cell lymphoma tumor. Eur J Dermatol. 2006;16:297-301.
-
35Khopkar U, Doshi BR, Dongre AM, Gujral S.A study of clinicopathologic profile of 15 cases of hypopigmented mycosis fungoides.Indian J Dermatol Venereol Leprol. 2011;77:167-73.
-
36Olsen EA, Whittaker S, Kim YH, Duvic M, Prince HM, Lessin SR, et al. Clinical end points and response criteria in mycosis fungoides and Sézary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011;29:2598-607.
-
37Wain EM, Orchard GE, Whittaker SJ, Spittle M Sc MF, Russell-Jones R.Outcome in 34 patients with juvenile-onset mycosis fungoides.Cancer. 2003;98:2282-90.
-
38El-Darouti MA, Marzouk SA, Azzam O, Fawzi MM, Abdel-Halim MR, Zayed AA, et al. Vitiligo vs. Hypopigmented mycosis fungoides (histopathological and immunohistochemical study, univariate analysis). Eur J Dermatol. 2006;16:17-22.
-
39Volkenandt M, Soyer HP, Cerroni L, Koch OM, Atzpodien J, Kerl H.Molecular detection of clone-specific DNA in hypopigmented lesions of a patient with early evolving mycosis fungoides.Br J Dermatol. 1993;128:423-8.
-
40Di Landro A, Marchesi L, Naldi L, Motta T, Cainelli T.A case of hypopigmented mycosis fungoides in a Young caucasian boy. Pediatr Dermatol. 1997;14:449-52.
-
41Breathnach SM, McKee PH, Smith NP.Hypopigmented mycosis fungoides: report of five cases with ultrastructural observations. Br J Dermatol. 1982;106:643-9.
-
42Bouloc A, Grange F, Delfau-Larue MH, Dieng MT, Tortel MC, Avril MF, et al. Leucoderma associated with flares of erythrodermic cutaneous T-cell lymphomas:four cases. Br J Dermatol. 2000;143:832-6.
-
43Chuang GS, Wasserman DI, Byers HR, Demierre MF.Hypopigmented T-cell discrasia evolving to hypopigmented mycosis fungoides during etanercept therapy.J Am AcadDermatol. 2008;59:S121-2.
-
44Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM,et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798-812.
-
45Moretti S, Spallanzani A, Amato L, Hautmann G, Gallerani I, Fabiani M, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92.
-
46Seif El Nasr H, Shaker OG, Fawzi MM, El-Hanafi G. Basic fibroblast growth factor and tumour necrosis factor alpha in vitiligo and other hypopigmented disorders: suggestive possible therapeutic targets. J EurAcadDermatolVenereol. 2013;27:103-8.
-
47Kitamura R, Tsukamoto K, Harada K, Shimizu A, Shimada S, Kobayashi T, et al. Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis: role of SCF/KIT protein interactions and the downstream effector, MITF-M. J Pathol. 2004;202:463-75.
-
48Duarte I, Bedrikow R, Aoki S. Mycosis fungoides: epidemiologic study of 17 cases and evaluation of PUVA photochemotherapy. An Bras Dermatol 2006;81:40-5
-
49Onsun N, Kural Y, Su O, Demirkesen C, Büyükbabani N.Hypopigmented mycosis fungoides associated with atopy in two children. PediatrDermatol. 2006;23:493-6.
-
50Kanokrungsee S, Rajatanavin N, Rutnin S, Vachiramon V.Efficacy of narrowband ultraviolet B twice weekly for hypopigmented mycosis fungoides in Asians.ClinExpDermatol. 2012;37:149-52.
-
*
Work performed at the Ambulatório de Oncologia Cutânea - Linfomas Cutâneos, da Divisão de Dermatologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP) - São Paulo, Brasil.
-
Financial Support: None
Publication Dates
-
Publication in this collection
Nov-Dec 2013
History
-
Received
03 Dec 2012 -
Accepted
14 Jan 2013